

Abstract
Dendritic epidermal T cells (DETCs) found in mouse skin are NKG2D-positive γδ T cells involved in immune surveillance and wound repair. It is assumed that the interaction of an NKG2D receptor on DETCs and an MHC class I-like NKG2D ligand on keratinocytes activates DETCs, which then secrete cytokines promoting wound repair. However, direct evidence that DETC activation through NKG2D signaling promotes wound repair is not available. In the present study, we generated mAbs for an NKG2D ligand H60c previously suggested to be expressed specifically on skin keratinocytes. Local administration of H60c-specific mAb inhibited activation of DETCs and significantly delayed wound repair. Likewise, administration of NKG2D-specific mAb impaired wound repair to a similar extent. The delay in wound closure resulting from the blockade of the NKG2D pathway was comparable to that observed in γδ T cell-deficient mice. These results indicate that H60c/NKG2D interactions play a critical role in wound repair. Reassessment of binding affinities showed that H60c monomers bind to NKG2D with affinity (Kd = 26 ± 3.2 nM) comparable to those of other high-affinity NKG2D ligands. H60c is transcribed not only in skin but also in tissues such as tongue and female reproductive tract known to contain epithelium-resident γδ T cells expressing invariant TCRs, suggesting a more general role for H60c in the maintenance of epithelial integrity.
In mice, γδ T cells constitute a minor subset of T lymphocytes in the blood and peripheral lymphoid tissues; however, they occur abundantly in epithelial layers of tissues such as skin, intestine, tongue, esophagus, trachea, lung, and reproductive tract (1–3). These epithelium-resident γδ T cells generally express TCRs with a very limited diversity (1, 4). Dendritic epidermal T cells (DETCs) are prototypic epithelium-resident γδ T cells distributed in the epidermal layer of the skin; they are predominantly derived from the “first wave” Vγ3+ fetal thymocytes and express an essentially monomorphic TCR consisting of Vγ3 (alternate nomenclature Vγ5) and Vδ1 chains, lacking junctional diversity (5). Unlike αβ T cells that recognize foreign Ag in the form of MHC-bound peptides through diverse TCRs, DETCs are thought to recognize injury- or stress-induced self-molecules in an MHC-unrestricted manner; activated DETCs secrete cytokines such as keratinocyte growth factors (KGFs) or kill stressed cells, thereby playing a crucial role in skin homeostasis and immune surveillance (3, 6, 7).
NKG2D ligands are a group of MHC class I-like molecules unique to mammals (8), the expression of which is induced by cellular stresses such as infection, tumorigenesis, heat shock, tissue damage, and DNA damage (9–11). Accumulated evidence indicates that the receptor for these ligands, NKG2D, is expressed by essentially all DETCs (12, 13), and that engagement of NKG2D ligands with NKG2D activates DETCs either by providing a costimulatory signal (13, 14) or without the need for TCR signaling (15). In mice, known NKG2D ligands include retinoic acid early inducible-1 (RAE-1)α–ε (16), histocompatibility (H60)a–c (17, 18), and mouse UL16-binding protein-like transcript 1 (MULT-1) (19, 20). Of these ligands, H60c identified in our laboratory is predominantly expressed in the skin (18), suggesting a role in skin immunity. Indeed, recent work demonstrated that H60c is a major NKG2D ligand gene whose expression is upregulated on cultured skin keratinocytes (presumably due to stress incurred by cell culture) and in damaged skin tissues, and that H60c delivers a costimulatory signal to DETCs, leading to the proposal that the interaction of H60c on keratinocytes and NKG2D on DETCs may be involved in wound repair and the maintenance of skin homeostasis (7, 14). However, direct evidence that DETC activation through NKG2D signaling facilitates wound repair is not available.
In the present study, we show that expression of H60c protein is induced on keratinocytes at the wound margin, and that blockade of interactions between H60c and NKG2D in vivo significantly impairs KGF secretion and wound repair. We also show that H60c is expressed constitutively in tissues other than skin, particularly in those tissues where epithelium-resident γδ T cells are abundant, suggesting a more general role for H60c in the maintenance of epithelial integrity.
Materials and Methods
Mice
TCRδ−/− mice, B6.129P2-Tcrdtm1Mom/J (stock no. 002120), were purchased from The Jackson Laboratory (Bar Harbor, ME). C57BL/6 mice, BALB/c Slc-nu/nu mice, and LEW/SsNSlc rats were from Sankyo Laboratory Service (Tokyo, Japan). TCRδ−/− mice and C57BL/6 mice were bred and housed under specific pathogen-free conditions in our animal facility. All experiments using animals were done according to the Guidelines for the Care and Use of Laboratory Animals at Hokkaido University Graduate School of Medicine and approved by the Institutional Review Committee of Hokkaido University.
Cell lines, Abs, and reagents
Ba/F3 cells expressing H60a, H60b, or H60c were generated as previously described (18) and maintained in complete RPMI 1640 medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% (v/v) heat-inactivated FBS, 10% (v/v) WEHI-3 cell-conditioned medium, and 3 μg/ml puromycin (Sigma-Aldrich). HEK293T cells were maintained in DMEM medium (Invitrogen, Carlsbad, CA) supplemented with 10% (v/v) heat-inactivated FBS at 37°C under 5% CO2. Rabbit anti-FLAG polyclonal Ab (F7425), rabbit IgG (I5006), and rat IgG (I8015) were purchased from Sigma-Aldrich. Hamster IgG was from Acris Antibodies (Herford, Germany). Rat IgG2a isotype control (14-4321) , rat IgG2b isotype control (14-4031) , rat anti-mouse RAE-1 mAb (MAB17581), and rat anti-mouse MULT-1 mAb (237104) were purchased from R&D Systems (Minneapolis, MN). FITC-conjugated hamster anti-mouse Vγ3 TCR mAb (553229), PE-conjugated hamster anti-mouse γδ TCR mAb (553178), and rat anti-mouse CD16/CD32 mAb (clone 2.4G2, 553141) were purchased from BD Biosciences (San Jose, CA). FITC-conjugated mouse anti-rat IgG (H+L) mAb (11-4811) and goat F(ab′)2 anti-rabbit polyclonal Ab (11-4839) used for FACS analysis were purchased from eBioscience (San Diego, CA). Peroxidase-conjugated goat anti-rat IgG (H+L) (14-16-12) for Western blotting and Alexa Fluor 488 goat anti-rat IgG (H+L) (A11006) for immunostaining were from KPL (Gaithersburg, MD) and Invitrogen, respectively. A blocking anti-NKG2D mAb, HMG2D, was described previously (21).
Preparation of H60c/Fc fusion protein
cDNAs encoding the α1 and α2 domains of H60c were generated from C57BL/6 mouse skin RNA by RT-PCR using the primers 5′-TCGATATCGGCCATGGGCACATATGCCCTAAATTGCA-3′ (sense) and 5′-GGCACTCCACAGATCTTGGCAAGGTTGGCATTTCTCT-3′ (antisense). The underlined sequences were included for in-frame insertion into the pFUSE-hIgG2-Fc2 vector (InvivoGen, San Diego, CA). This vector construct was transiently transfected into HEK293T cells, and the supernatants were collected; H60c/human IgG2 Fc fusion protein was purified on a HiTrap protein G HP column (GE Healthcare, Piscataway, NJ). Variable lymphocyte receptor (VLR)/Fc fusion protein (22) was prepared similarly and used as a control.
mAb production
Six- to 8-wk-old female LEW/SsNSlc rats were immunized once into footpads with 100 μg bacterially produced H60c protein (18) or H60c/Fc fusion protein mixed with TiterMax Gold adjuvant (CytRx, Los Angeles, CA). Lymphocytes isolated from popliteal lymph nodes were fused with myeloma cell line X63Ag8.653 using PEG1500. Hybridomas were injected into the peritoneal cavity of BALB/c Slc-nu/nu mice, and ascitic fluids were collected. Two mAbs against H60c, designated 1F10 (IgG2a subclass) and 5G6 (IgG2b subclass), were established; mAbs were purified using a HiTrap protein G HP column and dialyzed against PBS.
Cell surface labeling and Western blotting
Cell surface labeling of H60c protein was performed as previously described (23). Deglycosylation with PNGase F (New England Biolabs, Ipswich, MA) was performed as described (24). Proteins in sample buffer were run on SDS-PAGE and blotted to Hybond-P (GE Healthcare). H60c was detected with 5G6 or 1F10, followed by HRP-conjugated goat anti-rat IgG polyclonal Ab. Blots were visualized with ECL Advance (GE Healthcare).
Real-time and conventional RT-PCR
Expression of NKG2D ligand genes was analyzed by RT-PCR using an Applied Biosystems 7500 real-time PCR system (Applied Biosystems, Carlsbad, CA) as previously described (18). Briefly, total cellular RNA was prepared from snap-frozen tissues or DETCs, treated with DNase I, and converted to cDNA with the SuperScript III kit (Invitrogen). PCR was set up in triplicate on 25 ng cDNA using a Power SYBR Green PCR Master Mix kit (Applied Biosystems). Cycling conditions were 1 cycle at 50°C for 2 min and 1 cycle at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s and annealing/extension at 60°C for 1min. The primer sequences for H60b and H60c were previously described (18). The primer sequences were 5′-TGGACACTCACAAGACCATG-3′ and 5′-CCCAGGTGGCACTAGGAGT-3′ for pan-RAE-1; 5′-CAATGTCTCTGTCCTCGGAA-3′ and 5′-CTGAACACGTCTCAGGCACT-3′ for MULT-1; 5′-AACTGTTCCAGCCCCGAGCG-3′ and 5′-TCCAACTGCCACGGTCCTGA-3′ for KGF-1; 5′-TGTGCACTGGTACCAACTGA-3′ and 5′-CTTATGGAGATTTGTTTCAGC-3′ for Vγ3 TCR; 5′-TGGAAAGAATGTCTTGATTGTTGAA-3′ and 5′-AGCTTGCAACCTTAACCATTTTG-3′ for hypoxanthine phosphoribosyltransferase; and 5′-CGGCGACGACCCATTCGAAC-3′ and 5′-GAATCGAACCCTGATTCCCCGTC-3′ for 18S rRNA. Threshold cycle values (Ct) were determined using the sequence detection software version 1.3.1 (Applied Biosystems) and transformed to 2−ΔCt × 1000 for relative expression as previously described (25). The slope and y-intercept of standard curve lines in H60, pan–RAE-1, and MULT-1 were equivalent. Conventional RT-PCR was performed using GoTaq Master Mix (Promega, Madison, WI). Cycling conditions were 1 cycle at 95°C for 2 min, followed by 30 cycles of denaturation at 95°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s.
Flow cytometry
For cell surface staining, single-cell suspensions (1 × 106 cells) were washed with PBS (pH 7.4) supplemented with 1% FBS and stained for 60 min at room temperature with appropriately diluted primary Ab in PBS (pH 7.4) containing 0.1% NaN3. After washing, cells were incubated in 100 μl PBS (pH 7.4) containing 0.1% NaN3 with FITC-conjugated secondary Ab. Flow cytometry was done with a FACSCanto II (BD Biosciences). A minimum of 2 × 104 events were collected per sample and data were analyzed with CellQuest software (BD Biosciences). To examine whether anti-H60c mAbs were able to block binding of H60c to NKG2D, stably transfected Ba/F3 cells expressing H60c were treated with the mAbs, and then their ability to bind to recombinant mouse NKG2D/Fc fusion protein (139-NK; R&D Systems) was evaluated using FITC-conjugated anti-human IgG as secondary Ab.
Cytotoxicity assays
The ability of 5G6 to block functional H60c/NKG2D engagement was evaluated by cytotoxicity assays as previously described (18). Briefly, NK cells used as effectors were prepared from BALB/c splenocytes using anti-NK (DX5) microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). NK cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS and 100 ng/ml recombinant mouse IL-2 (R&D Systems) for 3 d before use. Prior to cytotoxicity assays, 106 NK cells were treated with 5 μg rat anti-mouse CD16/CD32 mAb for 20 min at 37°C. Cytotoxicity assays were performed for 4 h at 37°C at an E:T ratio of 10:1 with 8 × 103 target cells (Ba/F3 cells stably transfected with H60c) per well using the CytoTox 96 nonradioactive cytotoxicity assay kit (Promega). For blocking experiments, 106 target cells were pretreated with 10 μg 5G6 or rat IgG2b for 30 min at 37°C; effector cells were added leaving 5G6 in the wells. Specific lysis was calculated as specified by the kit.
Immunofluorescence staining
Ba/F3 cells stably expressing H60a, H60b, or H60c were attached on silane-coated glass slides by cytospin. Glass slides were fixed by cold acetone and washed twice with PBS; anti-H60c mAb 5G6 was added for 16 h at 4°C. Alexa Fluor 488 goat anti-rat IgG was then added for 30 min at room temperature, and cells were rinsed in PBS and mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Cells were observed with a fluorescence microscope.
Immunohistochemistry
Skin tissues were harvested at 1, 3, 6, and 12 h after wounding for immunohistochemical analysis. Frozen skin sections were prepared with CryoMount II (Muto Pure Chemicals, Tokyo, Japan) and fixed on slides with Cytosetter (Matsunami Glass, Osaka, Japan). After blocking with 10% goat serum for 1 h at room temperature, the sections were stained as described in the preceding paragraph.
Wound closure assay
TCRδ−/− and C57BL/6 mice were used between 8 and 12 wk of age. The dorsal surface of the mouse was shaved and left for 3 d. The pink-colored back skin and panniculus of anesthetized mice were pulled up, and two sets of sterile full-thickness wound were generated using a sterile 2-mm punch tool. Anti-H60c mAb (2 or 10 μg), anti-NKG2D mAb (10 μg), or 10 μg control Ig (cIg) was applied to each wound immediately after wounding as described (26). Wounds were left uncovered, and mice were housed individually with sterile wood chip bedding. Wounds on at least three mice were examined per condition in at least three independent experiments. Wounds were photographed daily for the following 6 or 7 d and wound areas were measured using ImageJ software (National Institutes of Health, Bethesda, MD).
Changes of DETC morphology at the wound margin
5G6 or normal rat IgG (200 μg each) was injected into the tail vein of C57BL/6 mice. Two hours after injection, mice were anesthetized, and a 2-mm hole was made in the center of both ears using a punch tool. Ears were collected at 3 and 6 h after wounding. Epidermal sheets were prepared as described previously (27). After incubation in 1% skim milk for 1 h at room temperature, the sheets were stained with FITC-conjugated anti-mouse Vγ3 TCR mAb for 16 h at 4°C. The proportion of DETCs that lost dendritic morphology was calculated by counting >300 cells at the wound margin at ×200 magnification. Counting was performed in two mice.
Preparation of DETCs from the wound margin
DETCs were prepared from wounded back skin as previously described (28). Briefly, a 2-mm border around the wound was collected at 12, 24, and 48 h after wounding. Epidermal sheets were prepared from the wound margin tissues by incubation with 5 mg/ml dispase for 3 h at 37°C. After enzymatic dissociation with DNase I, epidermal cells were filtered through a 70-μm mesh. After enrichment with Lympholyte-M (Cedarlane Laboratories, Hornby, ON, Canada), they were reacted with PE-conjugated anti-γδ TCR mAb, followed by sorting with anti-PE microbeads (Miltenyi Biotec). The purity of DETCs (>90%) was verified by FACS analysis using Vγ3 TCR mAb.
Preparation and characterization of recombinant H60c protein
Expression vectors for the N-terminal ectodomain of H60c were constructed as described previously (18). Each of the five cysteine residues of H60c was changed to serine by a PCR-based procedure. The forward primer sequences were 5′-TTCACGCATATGCTAAATAGCAAACTCACT-3′for the Cys20Ser mutant and 5′-TTCACGCATATGCTAAATTGCAAACTCACTGT-3′ (with an NdeI site underlined) for the other mutants. The reverse primer sequence was 5′-CCAAGCTTTTATCATCGAGGCTGGTCCACATGTG-3′ (with a HindIII site underlined). The PCR products were inserted into the pGMT7 vector using the NdeI and HindIII sites (29); the integrity of expression constructs was verified by sequencing. All recombinant proteins were expressed in Escherichia coli BL21 (DE3) pLysS strain as inclusion bodies and renatured by dilution refolding as previously described (18). The refolding mixtures were purified by gel filtration chromatography using HiLoad 26/60 Superdex 75 preparation grade (GE Healthcare) and subjected to SDS-PAGE with or without 10 mM DTT. After electrophoresis, gels were stained by Quick-CBB (Wako, Tokyo, Japan).
Determination of binding affinities
To measure the affinity of H60c to NKG2D, surface plasmon resonance analysis was performed as previously described (18) using Biacore 2000 (Biacore Life Sciences, Piscataway, NJ). Briefly, recombinant mouse NKG2D/Ig and BSA were coupled to a research-grade CM5 sensor chip via primary amines using a standard amine coupling kit (Biacore Life Sciences). H60c or BSA (as control soluble protein) was injected over the immobilized NKG2D and BSA at 25°C. The binding response at each concentration of H60c or at 1 μM BSA was calculated by subtracting the equilibrium response measured in the BSA flow cell from the response in the NKG2D flow cell. Dissociation constants were derived by nonlinear curve fitting of the standard Langmuir binding isotherm. Curve fitting was performed using the Origin 7 software (OriginLab, Northampton, MA).
Statistical analysis
The statistical significance of the data was determined by a Student t test. A p value <0.05 was taken as significant.
Results
Production of mAbs specific for H60c
We generated two mAbs specific for H60c and named them 5G6 and 1F10. Both mAbs reacted with H60c, but not H60a or H60b, when assessed by flow cytometry (Fig. 1A), Western blotting (Fig. 1B), and immunostaining (Fig. 1C). Pretreatment of H60c-expressing Ba/F3 cells with 5G6 blocked subsequent binding of NKG2D/Ig fusion protein (Fig. 1D), indicating that 5G6 blocks the binding of H60c to NKG2D. In contrast, similar treatment with 1F10 did not inhibit subsequent binding of NKG2D/Ig fusion protein (Fig. 1D), indicating that 1F10 binds to H60c, but lacks the ability to block H60c/NKG2D interactions. 5G6 significantly inhibited NK cell-mediated killing of Ba/F3 cells expressing H60c (Fig. 1E), demonstrating the ability of 5G6 to block functional H60c/NKG2D engagement.

Generation of H60c-specific mAbs. (A) Stably transfected Ba/F3 cells expressing H60a, H60b, or H60c (H60a-Ba/F3, H60b-Ba/F3, or H60c-Ba/F3) were subjected to flow cytometry after staining with 1F10, 5G6, anti-FLAG polyclonal Ab (open histograms), or isotype control Ab (filled histograms). (B) Recombinant H60 (rH60) proteins produced in E. coli were subjected to Western blot analysis using 5G6 as a probe. 1F10 was also specific for rH60c (data not shown). (C) Acetone-fixed Ba/F3 transfectant cells were stained with 5G6. Original magnification ×400. (D) Ba/F3 cells expressing H60c were pretreated with 1F10 or 5G6 and then reacted with NKG2D/Ig fusion protein (open histogram with a thick line) or VLR/Fc fusion protein (open histogram with a thin line). Anti-human IgG-FITC was used as secondary Ab. As a control, H60c-Ba/F3 cells were pretreated with an isotype control Ab and reacted with NKG2D-Ig fusion protein (filled histogram). (E) H60c-Ba/F3 cells were treated with 5G6 or rat IgG2b (cIg) and subjected to cytotoxicity assays using NK cells as effector cells at an E:T ratio of 10:1.
Generation of H60c-specific mAbs. (A) Stably transfected Ba/F3 cells expressing H60a, H60b, or H60c (H60a-Ba/F3, H60b-Ba/F3, or H60c-Ba/F3) were subjected to flow cytometry after staining with 1F10, 5G6, anti-FLAG polyclonal Ab (open histograms), or isotype control Ab (filled histograms). (B) Recombinant H60 (rH60) proteins produced in E. coli were subjected to Western blot analysis using 5G6 as a probe. 1F10 was also specific for rH60c (data not shown). (C) Acetone-fixed Ba/F3 transfectant cells were stained with 5G6. Original magnification ×400. (D) Ba/F3 cells expressing H60c were pretreated with 1F10 or 5G6 and then reacted with NKG2D/Ig fusion protein (open histogram with a thick line) or VLR/Fc fusion protein (open histogram with a thin line). Anti-human IgG-FITC was used as secondary Ab. As a control, H60c-Ba/F3 cells were pretreated with an isotype control Ab and reacted with NKG2D-Ig fusion protein (filled histogram). (E) H60c-Ba/F3 cells were treated with 5G6 or rat IgG2b (cIg) and subjected to cytotoxicity assays using NK cells as effector cells at an E:T ratio of 10:1.
H60c protein is expressed on keratinocytes at the wound margin
Previous work showed that H60c transcripts are detectable predominantly in the skin (18). More recently, it was shown that H60c is a major NKG2D ligand expressed on cultured keratinocytes, and that H60c mRNA levels are increased in wounded skin tissues (14). To examine whether keratinocytes at wound sites express H60c protein, we harvested the skin tissues at 12 h after wounding and performed immunofluorescence analysis using mAb 5G6 (Fig. 2A). Keratinocytes in the nonwounded area yielded only background fluorescence (Fig. 2A, top left panel). In contrast, keratinocytes at the wound margin yielded both cell surface and cytoplasmic staining (Fig. 2A, middle left panel). At higher magnification, cell surface staining was more evident. Peripheral to the wound margin was a transition zone where keratinocytes gradually lost staining with 5G6 (Fig. 2A, top right panel). Abs detecting RAE-1α, β, δ, and ε isoforms or MULT-1 did not stain keratinocytes at the wound margin (Fig. 2A, bottom left and right panels). These results indicate that expression of H60c, but not RAE-1 or MULT-1, is induced on stressed keratinocytes at the wound margin.

Expression of H60c protein in wounded skin. (A) Skin sections harvested at 12 h after wounding were stained with 5G6, cIg, Ab for RAE-1, or Ab for MULT-1 (original magnification, ×400). Ab for RAE-1 detects both δ and ε isoforms expressed in C57BL/6 mice. The image in the top left panel was obtained from the area ∼5 mm apart from the wound margin. The image in the top right panel shows the boundary region where keratinocytes proximal to the wound (left) are stained with 5G6, and those distal to the wound (right) are not stained. Insets show higher magnification images (digitally magnified ×2). Note that the stratum corneum yielded nonspecific fluorescence (as indicated by staining with cIg). (B) Skin sections harvested at 1, 3, 6, and 12 h after wounding were stained with 5G6 to examine the time course of H60c protein expression. Original magnification ×400.
Expression of H60c protein in wounded skin. (A) Skin sections harvested at 12 h after wounding were stained with 5G6, cIg, Ab for RAE-1, or Ab for MULT-1 (original magnification, ×400). Ab for RAE-1 detects both δ and ε isoforms expressed in C57BL/6 mice. The image in the top left panel was obtained from the area ∼5 mm apart from the wound margin. The image in the top right panel shows the boundary region where keratinocytes proximal to the wound (left) are stained with 5G6, and those distal to the wound (right) are not stained. Insets show higher magnification images (digitally magnified ×2). Note that the stratum corneum yielded nonspecific fluorescence (as indicated by staining with cIg). (B) Skin sections harvested at 1, 3, 6, and 12 h after wounding were stained with 5G6 to examine the time course of H60c protein expression. Original magnification ×400.
We then examined the time course of H60c protein expression after wounding (Fig. 2B). H60c protein was not clearly detectable in keratinocytes at 1 h after wounding. However, it became detectable by 3 h after wounding and its expression level was increased at 6 and 12 h after wounding.
Blockade of H60c/NKG2D interactions in vivo impairs wound repair
To evaluate the functional significance of H60c in wound repair, we examined whether blockade of H60c/NKG2D interactions could alter wound closure kinetics. Local administration of 10 μg 5G6 significantly delayed wound closure; compared with wounds treated with cIg, wounds treated with 5G6 required ∼1 more day to achieve comparable closure (Fig. 3A). This delay in wound closure was dose-dependent because administration of 2 μg 5G6 did not significantly delay wound closure (Fig. 3B). Furthermore, administration of 10 μg 1F10 with no blocking activity failed to alter wound closure kinetics (Fig. 3C), indicating that delayed wound repair results from the blockade of H60c/NKG2D engagement. Wound closure is known to be delayed in γδ T cell-deficient mice (30). Wounds treated with 5G6 exhibited repair kinetics similar to those seen in γδ T cell-deficient mice (Fig. 3D).
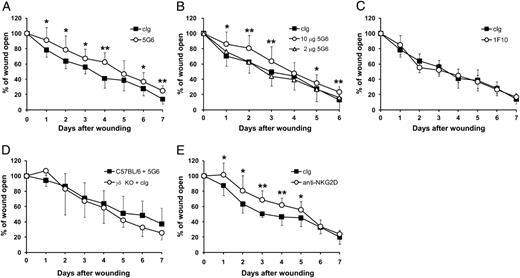
Blockade of H60c/NKG2D interactions impairs wound repair. Two sets of wounds were made on the back skin of C57BL/6 and TCRδ−/− mice. Immediately after wounding, 2 or 10 μg Abs was applied to each wound. (A) Wound closure in C57BL/6 mice treated with 10 μg 5G6 or cIg. (B) Wound closure in C57BL/6 mice treated with 2 or 10 μg 5G6 or 10 μg cIg. (C) Wound closure in C57BL/6 mice treated with 10 μg 1F10 or cIg. (D) Wound closure in C57BL/6 mice treated with 10 μg 5G6 and in TCRδ−/− mice treated with 10 μg cIg. (E) Wound closure in C57BL/6 mice treated with 10 μg HMG2D or cIg. cIgs for (A)–(D) were rat IgG. For (E), hamster IgG was used as a control. Data represent means ± SDs of 10–14 wounds per condition. All data are representative of three experiments. *p < 0.05, **p < 0.01 as determined by Student t test.
Blockade of H60c/NKG2D interactions impairs wound repair. Two sets of wounds were made on the back skin of C57BL/6 and TCRδ−/− mice. Immediately after wounding, 2 or 10 μg Abs was applied to each wound. (A) Wound closure in C57BL/6 mice treated with 10 μg 5G6 or cIg. (B) Wound closure in C57BL/6 mice treated with 2 or 10 μg 5G6 or 10 μg cIg. (C) Wound closure in C57BL/6 mice treated with 10 μg 1F10 or cIg. (D) Wound closure in C57BL/6 mice treated with 10 μg 5G6 and in TCRδ−/− mice treated with 10 μg cIg. (E) Wound closure in C57BL/6 mice treated with 10 μg HMG2D or cIg. cIgs for (A)–(D) were rat IgG. For (E), hamster IgG was used as a control. Data represent means ± SDs of 10–14 wounds per condition. All data are representative of three experiments. *p < 0.05, **p < 0.01 as determined by Student t test.
To examine whether blocking on the receptor side also impairs wound closure, we administered HMG2D, an NKG2D-specific blocking mAb, to the wound sites; this treatment exerted similar effects as the administration of 5G6 (Fig. 3E). These results indicate that interactions of NKG2D and its ligand H60c are indispensable for efficient wound closure.
Administration of H60c-specific mAb attenuates morphologic changes of DETCs in response to tissue damage and decreases the production of KGF-1 mRNAs
To confirm that the delayed wound closure described above involved the inhibition of DETC activation, we punched a hole in the mouse ear and examined the morphology of DETCs recovered from the wound margin (Fig. 4). DETCs normally have a characteristic dendritic shape; however, they become rounded when activated in response to tissue damage (30). Loss of dendritic morphology was evident at 3 h after wounding (Fig. 4A). When 5G6 was injected i.v. into mice 2 h before wounding, the proportion of DETCs that lost dendritic morphology at 3 and 6 h after wounding was decreased (Fig. 4A). This decrease was statistically significant when the number of DETCs with nondendritic morphology was compared between mice treated with 5G6 and those treated with cIg (Fig. 4B). These observations indicate that administration of 5G6 blocked activation of DETCs at wound sites.

Administration of 5G6 inhibits DETC activation. (A) cIg (rat IgG) or 5G6 was injected i.v. into mice 2 h before ear skin was wounded. At 3 and 6 h after injection, epidermal sheets were prepared from the ear skin and stained with Ab for mouse Vγ3. Red lines stand for the wound margin. Original magnifications are ×100 (top and middle panels) and ×400 (bottom panels). (B) DETCs at the wound margin were counted at original magnification ×200; the proportion of DETCs with dendritic morphology was calculated in mice treated with cIg (filled column) or 5G6 (open column). Results obtained in two mice were combined for statistical analysis. *p < 0.05. (C) Expression of KGF-1 mRNA, 18S rRNA, and Vγ3 mRNA. DETCs were isolated at the indicated time intervals from the wound margin in mice treated with cIg or 5G6.
Administration of 5G6 inhibits DETC activation. (A) cIg (rat IgG) or 5G6 was injected i.v. into mice 2 h before ear skin was wounded. At 3 and 6 h after injection, epidermal sheets were prepared from the ear skin and stained with Ab for mouse Vγ3. Red lines stand for the wound margin. Original magnifications are ×100 (top and middle panels) and ×400 (bottom panels). (B) DETCs at the wound margin were counted at original magnification ×200; the proportion of DETCs with dendritic morphology was calculated in mice treated with cIg (filled column) or 5G6 (open column). Results obtained in two mice were combined for statistical analysis. *p < 0.05. (C) Expression of KGF-1 mRNA, 18S rRNA, and Vγ3 mRNA. DETCs were isolated at the indicated time intervals from the wound margin in mice treated with cIg or 5G6.
Activated DETCs produce KGF-1, a cytokine promoting wound repair (31). Administration of 5G6 inhibited the production of KGF-1 mRNA in DETCs isolated from the wound site (Fig. 4C). This inhibition was evident at 12 and 24 h after administration of 5G6, but the KGF-1 mRNA levels were restored at 48 h, presumably because the blocking effects of 5G6 waned over time.
H60c monomers bind to NKG2D with affinity comparable to those of high-affinity NKG2D ligands
We previously showed that H60c binds to NKG2D with a Kd value of 8.7 ± 3.1 μM (18), thus with affinity considerably weaker than those of other NKG2D ligands such as MULT-1 or H60a (Kd = 6–30 nM) (19, 32). We subsequently noted that bacterially produced H60c protein is a dimer (Fig. 5A), whereas H60c protein produced in Ba/F3 cells occurs predominantly in a monomer (Fig. 5B), indicating that H60c dimers produced in bacteria most likely represent an artifact resulting from protein folding at very high concentrations. We therefore decided to measure the binding affinity of a monomeric form of H60c.

H60c monomer binds to NKG2D with affinity comparable to those of other high-affinity NKG2D ligands. (A) Bacterially produced H60a–c proteins were subjected to SDS-PAGE under nonreducing and reducing conditions. Open arrowhead indicates dimeric H60c; filled arrowhead indicates monomeric H60c. (B) Deglycosylated H60c protein expressed on Ba/F3 cells is a monomer. (C) Bacterially produced H60c protein C150S, in which cysteine at position 150 was changed to serine, occurs as a monomer (filled arrowhead), whereas bacterially produced wild-type H60c protein occurs as a dimer (open arrowhead). (D) Serially diluted H60c protein (from 35 to 0.5 μM) was injected through flow cells with immobilized NKG2D or BSA. The binding response at each concentration of H60c protein was calculated by subtracting the equilibrium response measured in the BSA flow cell from the response in the NKG2D flow cell. The solid line represents a direct nonlinear fit of the 1:1 Langmuir binding isotherm. Kd values with SDs were calculated from three independent experiments.
H60c monomer binds to NKG2D with affinity comparable to those of other high-affinity NKG2D ligands. (A) Bacterially produced H60a–c proteins were subjected to SDS-PAGE under nonreducing and reducing conditions. Open arrowhead indicates dimeric H60c; filled arrowhead indicates monomeric H60c. (B) Deglycosylated H60c protein expressed on Ba/F3 cells is a monomer. (C) Bacterially produced H60c protein C150S, in which cysteine at position 150 was changed to serine, occurs as a monomer (filled arrowhead), whereas bacterially produced wild-type H60c protein occurs as a dimer (open arrowhead). (D) Serially diluted H60c protein (from 35 to 0.5 μM) was injected through flow cells with immobilized NKG2D or BSA. The binding response at each concentration of H60c protein was calculated by subtracting the equilibrium response measured in the BSA flow cell from the response in the NKG2D flow cell. The solid line represents a direct nonlinear fit of the 1:1 Langmuir binding isotherm. Kd values with SDs were calculated from three independent experiments.
Bacterially produced H60c becomes a monomer when subjected to SDS-PAGE under reducing conditions, indicating the involvement of disulfide bridges in dimerization (Fig. 5A). H60c has cysteine residues at positions 20, 33, 56, 150, and 151 (18). To determine which cysteine residue is involved in dimerization, we generated a series of H60c mutant proteins in E. coli where each cysteine residue was substituted with a serine residue. Dimerization of H60c was abrogated only when cysteine at position 150 was substituted with serine (Fig. 5C). The monomeric form of bacterially produced H60c thus obtained (abbreviated as H60c C150S) bound to NKG2D with affinity (Kd = 26 ± 3.2 nM) comparable to those of other high-affinity NKG2D ligands (Fig. 5D).
H60c is expressed constitutively at low levels in tissues other than skin
A previous survey of representative organs and tissues by RT-PCR and Northern blotting suggested skin-specific expression of the H60c gene (14, 18). To examine whether expression of H60c is skin-specific, we performed more extensive tissue distribution analysis (Fig. 6A). This showed that, besides skin, H60c is expressed constitutively in tissues such as eyes, tongue, esophagus, vagina, and uterus. In these tissues, other NKG2D ligand genes were expressed at lower levels than H60c (Fig. 6B).

H60c transcripts are detected in tissues other than skin. (A) Expression of H60c was examined by real-time RT-PCR in the indicated tissues taken from adult healthy C57BL/6 mice. (B) H60c is a major NKG2D ligand gene expressed in eyes, tongue, esophagus, skin, and vagina when assessed by real-time RT-PCR. Raet1 and Ulbp1 stand for genes coding for RAE-1 and MULT-1, respectively. Raet1 gene expression was measured using primers amplifying all known isoform mRNAs. C57BL/6 mice do not express H60a (17, 18) because it is inactivated by a deletion (37). Hypoxanthine phosphoribosyltransferase was used as an internal control. For (A) and (B), data represent the mean of triplicate samples; error bars represent SDs.
H60c transcripts are detected in tissues other than skin. (A) Expression of H60c was examined by real-time RT-PCR in the indicated tissues taken from adult healthy C57BL/6 mice. (B) H60c is a major NKG2D ligand gene expressed in eyes, tongue, esophagus, skin, and vagina when assessed by real-time RT-PCR. Raet1 and Ulbp1 stand for genes coding for RAE-1 and MULT-1, respectively. Raet1 gene expression was measured using primers amplifying all known isoform mRNAs. C57BL/6 mice do not express H60a (17, 18) because it is inactivated by a deletion (37). Hypoxanthine phosphoribosyltransferase was used as an internal control. For (A) and (B), data represent the mean of triplicate samples; error bars represent SDs.
Discussion
DETCs play a crucial role in the maintenance of epidermal homeostasis and response to tissue damage, infection, inflammation, and malignancy (3, 7). Because all DETCs essentially express a single TCR made up of Vγ3 and Vδ1 chains with no junctional diversity, they are supposed to recognize a tissue-specific, restricted set of self-molecules whose expression is induced in situations of tissue dysregulation. However, the identity of self-molecules recognized by Vγ3-Vδ1 TCR remains unknown. A group of stress-inducible self-molecules generally assumed to function as a costimulatory signal for DETC activation are NKG2D ligands (13, 14, 33). H60c is a major NKG2D ligand expressed in skin (18). Thus, among known NKG2D ligands, H60c is clearly the most attractive candidate for a ligand involved in DETC activation. Indeed, in line with this idea, recent work showed that H60c mRNA is upregulated in wounded skin, and that keratinocytes expressing H60c are lysed by DETCs in vitro (14). However, direct evidence that H60c/NKG2D interactions are involved in wound repair was not available until now. In the present study, we demonstrated that expression of H60c protein is induced on keratinocytes at the wound margin (Fig. 2), and that blockade of H60c/NKG2D interactions impairs wound repair (Fig. 3) by inhibiting DETC activation (Fig. 4). Expression of H60c protein by 3 h after wounding (Fig. 2B) is consistent with the idea that this NKG2D ligand is involved in DETC activation that also occurred by 3 h after wounding (Fig. 4A).
Recently, coxsackie and adenovirus receptor (CAR) was identified as a costimulatory molecule involved in the activation of DETCs (26, 34). CAR, whose expression is induced on injured skin keratinocytes, binds to a membrane protein named junctional adhesion molecule-like protein (JAML) expressed on DETCs, leading to DETC activation. Blockade of the interaction between CAR and JAML impairs wound repair (26) in a manner comparable to the blockade of the interaction between H60c and NKG2D. Because H60c and CAR are both expressed on wounded keratinocytes and wound repair is impaired by the blockade of either H60c/NKG2D or CAR/JAML interactions, efficient wound repair appears to require both ligand/receptor pair interactions. There is some controversy as to whether NKG2D ligands deliver a costimulatory signal (13, 14) or whether their engagement with NKG2D alone is sufficient for DETC activation (15). The idea that efficient wound repair requires both H60c/NKG2D and CAR/JAML interactions favors the view that, under physiologic conditions, NKG2D ligand/receptor interactions alone are insufficient for optimal activation of DETCs.
H60c-expressing cells were lysed by NK cells as effectively as H60a- or H60b-expressing cells (18), despite the fact that H60c binds to NKG2D with affinity >10-fold weaker than those of any known NKG2D ligands, and >200- and 20-fold weaker than those of H60a and H60b, respectively (18). We found that H60 protein expressed in mammalian cells is a monomer whereas bacterially produced H60c protein used previously for affinity measurement is a dimer (Fig. 5). The monomeric form of H60c, which most likely represents a physiologic form, bound to NKG2D with affinity (Kd = 26 ± 3.2 nM) comparable to those of other high-affinity ligands such as H60a (Kd = 20–30 nM) (18, 32) and MULT-1 (Kd = 6 nM) (19). High binding affinity of H60c monomers is consistent with the observation that H60c exerts potent effects on wound repair kinetics (Fig. 3).
Interestingly, H60c is constitutively expressed in several tissues other than skin, particularly in eyes, tongue, esophagus, and female reproductive tract (Fig. 6). In the mouse fetus, shortly after the emigration of Vγ3-Vδ1 thymocytes to the epidermis where they differentiate into DETCs, thymocytes bearing an invariant Vγ4-Vδ1 TCR migrate to the epithelia of the tongue and female reproduction tract, where they are supposed to perform functions equivalent to those of DETCs (35). The corneal margin of the eye contains intraepithelial T cells, largely made up of γδ T cells, and corneal epithelial repair is impaired in TCRδ−/− mice (36). These observations suggest that H60c may regulate the activation of epithelium-resident γδ T cells not only in skin but also in other epithelial tissues, implicating a more general role for H60c in the maintenance of epithelial integrity.
Acknowledgements
We thank Dr. Yoichi Sutoh for VLR/Ig fusion protein and Chisato Sudo for technical assistance.
Footnotes
This work was supported by a Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research and by grants from the Northern Advancement Center for Science and Technology Foundation and the Uehara Memorial Foundation. R.H.M. is supported by a fellowship from the Egyptian government.
Abbreviations used in this article:
References
Disclosures
The authors have no financial conflicts of interest.
