

Abstract
Human papillomavirus (HPV) infection, particularly type 16, is causally associated with the development of cervical cancer. The E6 and E7 proteins of HPV are constitutively expressed in cervical carcinoma cells making them attractive targets for CTL-based immunotherapy. However, few studies have addressed whether cervical carcinomas can process and present HPV E6/E7-derived Ags for recognition by CTL. We generated HLA-A*0201-restricted CTL clones against HPV16 E629–38 that recognized HPV16 E6 Ags transfected into B lymphoblastoid cells. These CTL were unable to recognize HLA-A*0201+ HPV16 E6+ cervical carcinoma cell lines even when the level of endogenous HPV16 E6 in these cells was increased by transfection. This defect in presentation of HPV16 E629–38 correlated with low level expression of HLA class I, proteasome subunits low molecular mass protein 2 and 7, and the transporter proteins TAP1 and TAP2 in the cervical carcinoma cell lines. The expression of all of these proteins could be up-regulated by IFN-γ, but this was insufficient for CTL recognition unless the level of HPV16 E6 Ag was also increased by transfection. CTL recognition of the HPV16 E629–38 epitope in 721.174 B cells was dependent on TAP expression but independent of immunoproteasome expression. Collectively, these findings suggest that presentation of the HPV16 E629–38 epitope in cervical carcinoma cell lines is limited both by the level of TAP expression and by the low level or availability of the source HPV E6 oncoprotein. These observations place constraints on the use of this, and potentially other, HPV-derived CTL epitopes for the immunotherapy of cervical cancer.
Cervical cancer is the second commonest form of cancer in women worldwide with an estimated 400,000 new cases diagnosed annually (1). Research over the past decade has shown conclusively that infection with human papillomavirus (HPV)3foot;3075f3;10;ZPICKFOOT;> is the most significant risk factor in the etiology of cervical cancer (2). HPV DNA is detectable in 99.7% of cervical cancers worldwide (3) and is often integrated into the host genome (4). Of the 15 high risk HPV types isolated from cervical carcinomas, HPV16 is the most frequently detected, occurring in >50% of cervical cancers regardless of geographical origin (5).
The principal transforming proteins of high risk HPVs are E6 and E7, which block cell cycle exit in epithelial cells committed to differentiation (6, 7, 8), thereby allowing viral replication. The major cellular targets of E6 and E7 are the p53 (9) and retinoblastoma (10) cell proteins, respectively, which play a pivotal role in the negative regulation of cell growth. E6 and E7 are constitutively expressed in cervical cancer cells, and their continuous expression is required for maintenance of the transformed phenotype (11, 12). These viral tumor Ags, which are not present in normal cells, make attractive targets for specific CD8+ CTL-based immunotherapy.
CTL recognize short peptides, 8–10 amino acids in length, presented on the surface of target cells in the context of MHC class I molecules (also known as HLA molecules in humans). These peptides are generated from full-length (viral) proteins by a complex intracellular processing pathway involving many components including the proteasomes and TAP complex (13). Candidate human CTL epitopes from HPV16 E6 and E7, restricted by the most frequent HLA-A molecules, including HLA-A*0201, have been predicted by HLA binding studies (14). In follow-up studies, three high-affinity (E711–20, E782–90, and E786–93) and one intermediate affinity (E629–38) binding peptides induced peptide-specific CTL responses in immunized HLA-A*0201/Kb transgenic mice (15). Primary human CTL responses were most frequently found against the three high affinity peptides and CTL clones induced against these peptides could kill a HLA-A*0201+ HPV16+ cervical carcinoma cell line (CaSki), suggesting that these peptides represent naturally processed human CTL epitopes of HPV16. In support of this CTL responses against two of the peptides (HPV16 E711–20 and E786–93) have been detected in individuals with cervical neoplasia (16, 17) but with variable efficiency (18). This may reflect the low frequency of HPV-specific CTL in blood (19).
Alternatively, the HPV peptide epitopes predicted by binding studies may not be the most immunogenic peptides in vivo, and other as yet unidentified peptides may be more effective at inducing HPV-specific immune responses. With this in mind, we attempted to define novel peptide epitopes within HPV16 E6 and E7, which could be targets for specific CTL, by using dendritic cells (DCs) as APCs in vitro. These DCs were infected with a recombinant adenovirus (rAd101) to express HPV16 E6 and E7 intracellularly, and used to stimulate a primary HLA-A*0201-restricted CTL response from the blood of a healthy donor. The epitope specificity of the HPV-specific CTL line generated was mapped, and the presentation of this epitope by HPV16+ cervical carcinomas was investigated.
Materials and Methods
Subjects
The study was approved by the South Glamorgan local ethics committee, and informed consent was obtained for all samples. Cervical cancer patients were recruited from patients presenting for radical treatment at the University Hospital of Wales (Cardiff, U.K.). Normal controls were laboratory staff with no known history of cervical dysplasia or neoplasia. PBMC from patients and controls were screened for HLA-A*0201 expression using the mAbs MA2.1 (HLA-A2 and HLA-B17; Ref. 20) and CR351-11 (recognizing HLA-A*0201 and HLA-A28; One Lambda, Canoga Park, CA). Blood from laboratory staff who were not HLA-A*0201 positive was used as a source of allogeneic feeder cells for use in cloning and expansion protocols.
Cell lines
The CIR.A2 cell line is a B lymphoblastoid cell line (B-LCL) expressing a transfected genomic clone of HLA-A*0201 (21). It was grown in RPMI/FCS (RPMI 1640 with 10% FCS, 2 mM glutamine, 100 μg/ml streptomycin, 100 U/ml penicillin, and 25 mM HEPES) and 400 μg/ml G418 (Life Technologies, Grand Island, NY). LCL721.174 (.174) cells have only one copy of chromosome 6 containing a deletion in the class II region of the MHC locus and therefore lack the TAP and low molecular mass protein (LMP2/7) genes (22, 23). The .174/TAP cells are .174 cells transfected with plasmids containing the TAP1 and TAP2 genes (24) and .174/TAPs/LMP7 are .174/TAPs cells transfected with a plasmid containing the LMP7b gene (25). They were grown in RPMI/FCS, and 500 μg/ml G418 (.174/TAPs and .174/TAPs/LMP7) and 200 μg/ml hygromycin (.174/TAPs/LMP7 only) were added to the transfected cell lines.
CaSki (American Type Culture Collection (ATCC; Manassas, VA) CRL-1550) is a HLA-A2, HPV16-transformed cervical carcinoma cell line derived from a small bowel metastasis (26). C33A-HPV16 is derived from a HLA-A2, HPV-negative cervical cancer cell line (C33A) transfected with a recombinant plasmid containing the HPV16 genome (27) (gift from Professor J. Dillner, Karolinska Institute, Stockholm, Sweden). SiHa (ATCC HTB-35) is a HLA-A2-negative, HPV16-transformed cervical carcinoma cell line derived from a squamous carcinoma of the cervix (28). MS751 (ATCC HTB-34) is a HLA-A2-positive, HPV18-transformed cervical carcinoma cell line established from a lymph node metastasis (29). MDA231 is a HLA-A2-positive, (HPV-negative) breast carcinoma cell line isolated from a pleural effusion (30) (gift from L. Sherman, Scripps Clinic, La Jolla, CA). CaSki and MDA231 were maintained in RPMI/FCS, whereas all other tumor cell lines were grown in DMEM/FCS (DMEM with 10% FCS, 2 mM glutamine, 100 μg/ml streptomycin, 100 U/ml penicillin, 25 mM HEPES, and 1% nonessential amino acids).
Viruses and synthetic peptides
RAd101 is a replication-deficient recombinant adenovirus containing a HPV16 E6/E7 fusion protein and has previously been used to stimulate HPV-specific CTL in vitro (31, 32). TA-HPV is a recombinant vaccinia virus expressing HPV16 and HPV18 E6/E7 fusion gene products, and Wyeth is the parental strain of vaccinia used in the construction of TA-HPV (33). SR16 and SR18 are recombinant vaccinia viruses expressing HPV16 E6/E7 gene products and HPV18 E6/E7 gene products, respectively (C. Boswell and J. Hickling, unpublished observations). All vaccinia viruses were provided by Cantab Pharmaceuticals (Cambridge, U.K.).
In vitro CTL induction using DCs
DCs were generated as previously described (34) with some modifications. PBMC were isolated from heparinized peripheral venous blood by Ficoll-Hypaque (Histopaque-1077; Sigma, St. Louis, MO) density gradient centrifugation, resuspended at 4 × 106 cells/ml in serum-free RPMI 1640, and distributed into six-well tissue culture plates (Greiner, Frickenhausen, Germany) at 3 ml/well. After 2 h at 37°C, nonadherent cells were gently removed, and 3 ml/well RPMI/AB (RPMI 1640 with 10% pooled human AB serum, 2 mM glutamine, 100 μg/ml streptomycin, 100 U/ml penicillin, and 25 mM HEPES) supplemented with 800 U/ml human rGM-CSF (Schering-Plough, Kenilworth, NJ) and 500 U/ml human rIL-4 (BD PharMingen, San Diego, CA) was added. The wells were fed every other day with fresh medium containing rGM-CSF and rIL4, and on day 5, 50 U/ml human rIL-1 (34, 35) was added. After 6 days, the DCs were harvested by gentle pipetting, resuspended in 200 μl of serum-free RPMI 1640, and infected with rAd101 at a multiplicity of infection of 200 PFU/cell for 2 h at 37°C. Thereafter, the infected DCs were washed twice, irradiated (5000 rad), and distributed at 1 × 105 cells/well into a 24-well plate in 1 ml/well RPMI/AB.
Ab detection of intracellular HPV E6/E7 expression is difficult (36); therefore, RT-PCR was used to confirm successful expression of HPV16 E7 in rAd101-infected DCs and monocytes (data not shown). Furthermore, other molecules such as HLA-A2, for which high affinity mAbs exist, could be detected at high levels in DCs after infection with a recombinant adenovirus encoding the HLA-A2 gene (our unpublished observations).
Autologous CD8+-enriched responder T lymphocytes were prepared from the nonadherent cell fraction obtained on day 0 by immunomagnetic beads (Dynal Biotech, Wirral, U.K.) and depletion of CD4+ and CD16+ cells (37), and then cryopreserved. On day 6, the cells were thawed and added at 2 × 106 cells/well to the adenovirus-infected DCs (for a responder-stimulator ratio of 20:1) in 1 ml/well RPMI/AB supplemented with human rIL-7 (Genzyme, Cambridge, MA) at a final concentration of 5 ng/ml.
After 10 days and weekly thereafter, the responder population was restimulated with rAd101-infected monocytes. On the day before restimulation, PBMCs were resuspended at 4 × 106/ml in serum-free RPMI 1640 and distributed into a 24-well plate at 1 ml/well. After 2 h at 37°C, nonadherent cells were discarded and adherent monocytes were washed and then infected with rAd101 at a multiplicity of infection of 2000 PFU/cell (as determined by titration on CV-1 indicator cells). After 1 h at 37°C, 1 ml/well RPMI/AB was added and incubated overnight at 37°C. The next day, the cells were washed to remove excess virus, and responders were added at 1–2 × 106 cells/well in RPMI/AB. One day later, 20 U/ml human rIL-2 (Chiron, Harefield, U.K.) was added to the wells. Responder populations were tested for their specificity in a standard 51Cr-release assay after three rounds of restimulation.
In vitro CTL induction using peptide stimulation
A modified version of a previously described method (38) was used. PBMC were resuspended at high cell density in a minimal volume (∼100 μl) of serum-free RPMI 1640 and pulsed at a peptide concentration of 100 μg/ml for 1 h at 37°C. Thereafter, the cells were diluted to 2 × 106 cells/ml with RPMI/AB and distributed at 1 ml/well into a 24-well plate with 20 ng/ml rIL-7. After 3 days, 10 U/ml rIL-2 was added to the wells. On day 7 and weekly thereafter, the responder cells were restimulated with autologous, peptide-pulsed, irradiated (5000 rad) PBMC at a responder-stimulator ratio of 2:1, and 10 U/ml IL-2 was added to the wells 3 days later. Responder populations were tested for their specificity in a 51Cr-release assay after one and/or two rounds of restimulation.
Cloning of T cell lines
HPV-specific CTL lines were seeded at 0.5–1 cell/well into 96-well plates in 150 μl/well RPMI/AB containing fresh, irradiated (5000 rad), allogeneic PBMC from three HLA-mismatched donors (5 × 104-1 × 105/well), irradiated allogeneic B-LCL (1 × 104/well) (optional), IL-2 (25 U/ml), and anti-CD3 mAb (0.03 μg/ml) (R&D Systems, Minneapolis, MN) (39). In some cloning procedures, PHA (Murex, Norcross, GA) (5 μg/ml on day 0, 1 μg/ml thereafter) (40) was used instead of anti-CD3 to stimulate T cell proliferation, and IL-2 was omitted until day 3. After 7 days, 50 μl of RPMI/AB containing IL-2 at a final concentration of 25 U/ml was added to the wells. On day 14, and weekly thereafter, the cultures were restimulated with RPMI/AB containing allogeneic PBMC, B-LCL, IL-2, and anti-CD3 mAb (or PHA) in the same concentrations as used on the first day of cloning. Cultures were assayed for specific cytotoxicity after 21 and/or 28 days, and positive wells were maintained in culture. Positive cultures were stained using a panel of anti-TCRBV mAbs (41) and analyzed on a FACS (FACSCalibur; BD Biosciences, Mountain View, CA). CTL that were CD8+ and expressed a single TCRBV chain were categorized as clones. Although we had no formal genetic proof for clonality these CTL clones maintained their phenotype and functional properties after extended tissue culture.
Cloned CTL (1 × 106) were expanded to large numbers in tissue culture flasks containing 50 ml of RPMI/AB with 20 × 106 fresh irradiated (5000 rad) allogeneic PBMC from three donors, IL-2 (20 U/ml), and anti-CD3 mAb (0.03 μg/ml) or PHA (1 μg/ml). Fresh medium and IL-2 were added on day 5. On day 7, the CTL were transferred to a 24-well plate at 2 × 106 cells/well and cultured in the presence of 100 U/ml IL-2 (42) for up to 14 days.
51Cr release cytotoxicity assays
When target cells were IFN-γ-treated, the IFN-γ (Roche, Basel, Switzerland) was added to culture medium at 200 U/ml for 48 h before cytotoxicity assays. Target cells were infected overnight with recombinant vaccinia at 15 PFU/cell at 37°C, before labeling with 51Cr (Amersham, Little Chalfont, U.K.). Peptide-pulsed target cells were incubated with peptide (10 μg/ml) for 1 h after 51Cr labeling, and washed twice before use. 51Cr-labeled target cells (2 × 103) were then added to triplicate wells of 2-fold serially diluted effector cells. Supernatants were harvested after 4 h. Percentage of specific lysis was calculated as [(mean experimental release − mean spontaneous release)/(mean maximum release − mean spontaneous release)] × 100. A CTL response was defined as an increase in specific lysis of at least 10% above controls at two or more E:T ratios (43).
Western blotting
Lysates were prepared from carcinoma cell lines by incubating with Nonidet P-40 lysis buffer (0.5% Nonidet P-40 detergent, 10% glycerol, 250 mM NaCl, 2 mM EDTA, 50 mM HEPES) on ice for 30–60 min. Thereafter, the samples were centrifuged and the supernatant was mixed with an equal volume of 2× gel sample buffer (125 mM Tris-HCl (pH 6.8), 20% glycerol, 10% 2-ME, 4% SDS, 0.004% bromophenol blue) and heated at 100°C for 3 min.
The cell lysates were resolved by SDS-PAGE on a 12% polyacrylamide gel and transferred to a polyvinylidine difluoride support membrane that was placed overnight in a casein-based blocking solution (I-Block; Tropix, Bedford, MA). The membranes were probed with rabbit anti-peptide sera (provided by Prof. J. Trowsdale and Dr. A. Kelly, Department of Immunology, Cambridge University, Cambridge, U.K.) specific for the proteasome subunits δ, mb1, LMP2, and LMP7, the transporter proteins TAP1 and TAP2, and the proteasomal regulator PA28 (44) diluted 1/1000 in blocking buffer. The membranes were washed extensively and then incubated with goat anti-rabbit alkaline-phosphatase-conjugated secondary Ab (Bio-Rad, Hercules, CA) diluted 1/10,000 in blocking buffer. The membranes were washed again and treated with CDP-Star (Tropix) for 10–20 min and then exposed to x-ray film for autofluorography.
Results
HPV16 E629–38-specific CTL recognize HPV16 E6 Ags processed and presented by B-LCL
Autologous DCs infected with a recombinant adenovirus expressing HPV16 E6/E7 (rAd101) were used to stimulate PBMC from a HLA-A*0201 healthy female volunteer. After three further stimulations with rAd101-infected monocytes, a CTL line was obtained that was able to recognize a HLA-A*0201-transfected B-LCL (C1R.A2) infected with a recombinant vaccinia virus (TA-HPV) expressing HPV16 and HPV18 E6/E7 (Fig. 1 A). As might be expected for a polyclonal CTL line, there were also lower levels of specific lysis of parental strain vaccinia (Wyeth)-infected or uninfected B-LCL targets.

HPV specific CTL can be generated in vitro using recombinant adenovirus-infected DCs. A, PBMC from a HLA-A*0201 healthy volunteer were stimulated with rAd101-infected DCs and assayed on day 35 against 51Cr-labeled C1R. A2 cells that were: uninfected (□), infected with a recombinant vaccinia virus containing HPV16 and HPV18 E6/E7 (TA-HPV) (•), or the parental Wyeth vaccinia expressing no HPV Ags (○). B, The HPV-specific CTL line was cloned by limiting dilution and a putative clone of CD8+ T cells (3C11) was assayed against 51Cr-labeled C1R. A2 cells that were either uninfected (□), infected with TA-HPV (•), Wyeth (○), a vaccinia virus encoding HPV16 E6/E7, SR16 (▵), or a vaccinia virus encoding HPV18 E6/E7, SR18 (▿). C, Clone 3C11 was assayed against 51Cr-labeled C1R. A2 cells that were unpulsed (□) or pulsed with 10 μg/ml the following HLA-A*0201 binding peptides: HPV16 E711–20 (┌), HPV16 E786–93 (✳), HPV16 E782–90 (┘), and HPV16 E629–38 (▴). D, Clone 7E7 was assayed against 51Cr-labeled C1R. A2 cells that were unpulsed (□) or infected with TA-HPV (•) or Wyeth (○), or pulsed with HPV16 E629–38 (▴) peptide.
HPV specific CTL can be generated in vitro using recombinant adenovirus-infected DCs. A, PBMC from a HLA-A*0201 healthy volunteer were stimulated with rAd101-infected DCs and assayed on day 35 against 51Cr-labeled C1R. A2 cells that were: uninfected (□), infected with a recombinant vaccinia virus containing HPV16 and HPV18 E6/E7 (TA-HPV) (•), or the parental Wyeth vaccinia expressing no HPV Ags (○). B, The HPV-specific CTL line was cloned by limiting dilution and a putative clone of CD8+ T cells (3C11) was assayed against 51Cr-labeled C1R. A2 cells that were either uninfected (□), infected with TA-HPV (•), Wyeth (○), a vaccinia virus encoding HPV16 E6/E7, SR16 (▵), or a vaccinia virus encoding HPV18 E6/E7, SR18 (▿). C, Clone 3C11 was assayed against 51Cr-labeled C1R. A2 cells that were unpulsed (□) or pulsed with 10 μg/ml the following HLA-A*0201 binding peptides: HPV16 E711–20 (┌), HPV16 E786–93 (✳), HPV16 E782–90 (┘), and HPV16 E629–38 (▴). D, Clone 7E7 was assayed against 51Cr-labeled C1R. A2 cells that were unpulsed (□) or infected with TA-HPV (•) or Wyeth (○), or pulsed with HPV16 E629–38 (▴) peptide.
This CTL line, which contained 53% CD8+ T lymphocytes (data not shown) was cloned by limiting dilution. This resulted in 18 clones of which only 1 was specific for TA-HPV-infected targets (>60% specific lysis, data not shown). This CTL clone, 3C11, was CD8+, TCRVB3+ and demonstrated improved specificity (Fig. 1,B) compared with the polyclonal CTL line (Fig. 1,A), with greater specific lysis of TA-HPV-infected targets at lower E:T ratios and negligible lysis of Wyeth-infected and uninfected targets. The CTL line was stimulated with HPV16 E6/E7 Ags, but tested on TA-HPV, which encoded both HPV16 E6/E7 and HPV18 E6/E7. The specificity of 3C11 for HPV16 E6/E7 was confirmed by recognition of targets infected with HPV16 E6/E7 vaccinia viruses (SR16) but not those infected with HPV18 E6/E7 vaccinia viruses (SR18) (Fig. 1 B).
HPV16 E629–38 peptide-specific CTL can be generated from patients with cervical cancer
The results described above demonstrate that CTL against HPV16 E629–38 could be generated from a healthy donor by stimulating T cells with DCs expressing full-length E6 proteins. Although the HPV status of this donor was not known, it was likely that this CTL response represented a primary in vitro response as we were unable to detect any T cells in PBMC using an HLA-A*0201/HPV16 E629–38 tetramer (data not shown). The possibility that “memory” T cell responses against HPV16 E629–38 could be detected in patients with cervical cancer was investigated. Using a short-term in vitro peptide restimulation protocol, significant CTL responses against HPV16 E629–38 were detected in the PBMC of 2 of 10 cervical cancer patients but not in 10 healthy volunteers (data not shown). From one patient a CD8+TCRBV8+ T cell clone (7E7) was generated by limiting dilution cloning. This CTL clone recognized both peptide-pulsed C1R.A2 cells as well as C1R.A2 cells expressing HPV16 E6 endogenously after infection with TA-HPV (Fig. 1 D).
Therefore, CTL generated using a “primary” whole Ag restimulation protocol (3C11) and a memory peptide restimulation protocol (7E7) have similar specificities; both recognize the HPV16 E629–38 epitope either provided exogenously as a synthetic peptide or after endogenous processing of whole HPV16 E6 Ags in B-LCL.
HPV16 E629–38 peptide-specific CTL do not recognize HLA-A*0201 HPV16 positive cervical carcinoma cells expressing the HPV16 E6 protein endogenously
To investigate the immunotherapeutic potential of HPV16 E629–38- specific CTL, clones 3C11 and 7E7 were tested against two HLA-A*0201+ cervical carcinoma cell lines: CaSki and C33A-HPV16. Caski is a cervical carcinoma that has been naturally transformed by HPV16 and expresses both E6 and E7 gene products. C33A-HV16 is also a cervical carcinoma but derived from an HPV-negative parental cell (C33A) that has subsequently been transfected with HPV16 E6 and E7 genes. Despite expression of HPV16 E6, neither cervical carcinoma cell line was recognized by 3C11 (Fig. 2 A) or by 7E7 (data not shown).
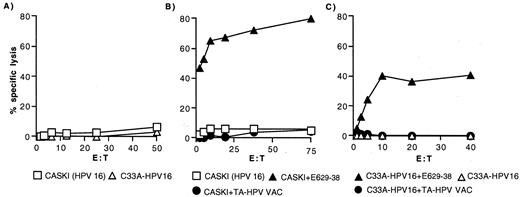
HLA-A*0201 HPV16-positive cervical carcinoma cells expressing the HPV16 E6 protein endogenously fail to present the HPV16 E629–38 epitope. A, Clone 3C11 was assayed against 51Cr-labeled CaSki (□) and C33A-HPV16 (▵) cells. Similar results were obtained with 7E7. B, Clone 3C11 was assayed against 51Cr-labeled CaSki cells that were: uninfected (□), infected with TA-HPV (•), or pulsed with HPV16 E629–38 (▴). Similar results were obtained with 7E7. C, Clone 3C11 was assayed against 51Cr-labeled C33A-HPV16 cells that were uninfected (▵), infected with TA-HPV (•), or pulsed with HPV16 E629–38 (▴). Similar results were seen with 7E7.
HLA-A*0201 HPV16-positive cervical carcinoma cells expressing the HPV16 E6 protein endogenously fail to present the HPV16 E629–38 epitope. A, Clone 3C11 was assayed against 51Cr-labeled CaSki (□) and C33A-HPV16 (▵) cells. Similar results were obtained with 7E7. B, Clone 3C11 was assayed against 51Cr-labeled CaSki cells that were: uninfected (□), infected with TA-HPV (•), or pulsed with HPV16 E629–38 (▴). Similar results were obtained with 7E7. C, Clone 3C11 was assayed against 51Cr-labeled C33A-HPV16 cells that were uninfected (▵), infected with TA-HPV (•), or pulsed with HPV16 E629–38 (▴). Similar results were seen with 7E7.
To investigate the possibility that there may be insufficient levels of HPV16 E6 within these cells for generation of the HPV16 E629–38 peptide, they were infected with TA-HPV but this did not reconstitute recognition (Fig. 2, B and C). In contrast, as previously shown, C1R.A2 B-LCL cells were recognized after infection with TA-HPV (Fig. 1, B and D). Both CaSki and C33A-HPV16 cells could be lysed by the HPV16 E629–38-specific CTL if the HPV16 E629–38 peptide was supplied exogenously (Fig. 2, B and C).
These results suggest an intracellular defect in CaSki and C33a-HPV16 cells affecting presentation of HPV16 E629–38, even when an excess of endogenous HPV16 E6 was provided by TA-HPV infection. This defect was not limited to the HPV16 E629–38 epitope, as CaSki cells also failed to present the HLA-A*0201-restricted influenza M158–66 epitope to M158–66-specific CTL after being infected with a recombinant vaccinia virus encoding M1 (data not shown).
Presentation of HPV16 E629–38 to CTL varies between HLA-A*0201+ carcinoma cell lines
To investigate whether these findings were peculiar to HPV16-positive cervical carcinoma cells, HPV16 E629–38-specific CTL were assayed against various HLA-A*0201 carcinoma cell lines, including those of cervical origin (MS751, C33A) and noncervical (MDA-231). Because these carcinoma cell lines do not normally express HPV16 Ags, they were all infected with TA-HPV as a source of endogenous E6 before being assayed for CTL recognition. Similar to the results with CaSki and C33A-HPV16, two cervical cell lines, MS751 (HPV18-transformed cervical cell line) and the C33A (HPV-negative parent of C33A-HPV16), failed to present the HPV16 E629–38 epitope after TA-HPV infection (Fig. 3, A and B). By contrast, the breast carcinoma cell line MDA231 (Fig. 3 C) was able to present the HPV16 E629–38 epitope efficiently.
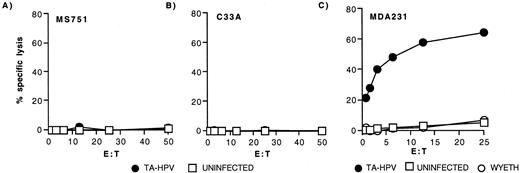
Variation among HLA-A*0201+ carcinoma cells in processing and presentation of the HPV16 E629–38 epitope. Clone 3C11 was assayed against 51Cr-labeled HPV18+ MS751 cells (A) and HPV-negative C33A cells (B) and MDA231 breast carcinoma cells (C) that were uninfected (□) or infected with TA-HPV (•). Both MS751 and C33A cells were also tested in the presence of HPV16 E629–38 peptide, with specific lysis of 69 and 37%, respectively, at the top E:T.
Variation among HLA-A*0201+ carcinoma cells in processing and presentation of the HPV16 E629–38 epitope. Clone 3C11 was assayed against 51Cr-labeled HPV18+ MS751 cells (A) and HPV-negative C33A cells (B) and MDA231 breast carcinoma cells (C) that were uninfected (□) or infected with TA-HPV (•). Both MS751 and C33A cells were also tested in the presence of HPV16 E629–38 peptide, with specific lysis of 69 and 37%, respectively, at the top E:T.
Cervical carcinoma cell lines express low or intermediate levels of cell surface MHC class I molecules, which are up-regulated by IFN-γ
There have been several reports of HLA class I down-regulation or loss in cervical carcinomas; therefore, the levels of cell surface HLA-A2.1 expression of the cervical carcinoma cell lines were determined (Fig. 4). In general, the level of HLA-A2.1 expression by the carcinoma cell lines was lower than that seen on B-LCL but there was a range of expression. The highest levels of HLA-A2 expression were seen in MDA231 cells, intermediate levels in CaSki and MS751 cells, and very low levels in C33A and C33A-HPV16 cells.
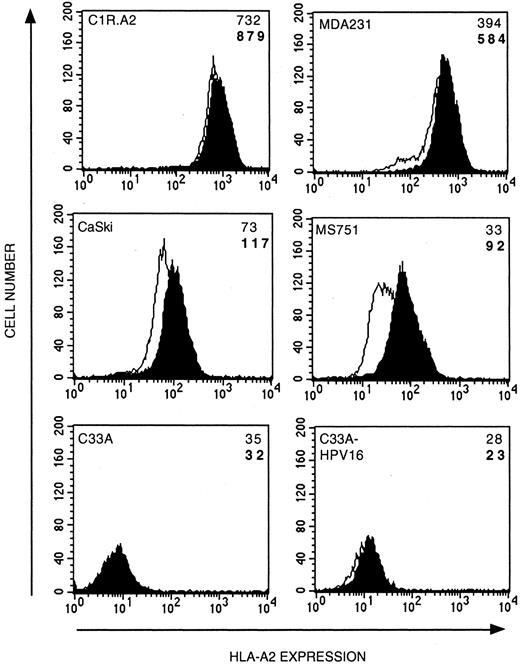
Cervical carcinoma cell lines express low or intermediate levels of cell surface MHC class I molecules that can be up-regulated by IFN-γ. Carcinoma cell lines cultured with 200 U/ml IFN-γ for 48 h (shaded) or left untreated (open) were analyzed for HLA-A*201 expression. The mean fluorescence intensity (MFI) for each result in the presence (bold font) or absence (normal font) of IFN-γ is shown. Mean fluorescence intensity for negative controls (FITC Ab only) was between 2 and 4.
Cervical carcinoma cell lines express low or intermediate levels of cell surface MHC class I molecules that can be up-regulated by IFN-γ. Carcinoma cell lines cultured with 200 U/ml IFN-γ for 48 h (shaded) or left untreated (open) were analyzed for HLA-A*201 expression. The mean fluorescence intensity (MFI) for each result in the presence (bold font) or absence (normal font) of IFN-γ is shown. Mean fluorescence intensity for negative controls (FITC Ab only) was between 2 and 4.
It is well established that IFNs up-regulate the expression of many proteins involved in the MHC class I Ag presentation pathway (45). Pretreatment with IFN-γ up-regulated HLA-A2.1 expression in all of the cell lines except C33A-HPV16 and C33A. HLA-A2.1 expression was up-regulated between 48% (MDA231) and 178% (MS751) (Fig. 4). The failure of IFN-γ to up-regulate the expression of HLA-A2.1 in the parental C33A as well as the transfectant C33A-HPV16 cells implies that IFN-γ nonresponsiveness was not a result of HPV16 transfection.
Cervical carcinoma cell lines express low levels of immunoproteasomes and TAP proteins, which can be up-regulated by IFN-γ
The processing and presentation of endogenous viral Ags for CD8 T cells is critically dependent on the expression of intracellular proteins such as TAP and the proteasomes. We determined the expression levels of these proteins in carcinoma cell lines by Western blot. In addition to the cell lines tested so far, the HLA-A*0201-negative HPV16-positive cervical carcinoma cell line SiHa was included.
The constitutive proteasome subunits δ and mb1 were expressed in every cell line analyzed, and their expression was not affected by IFN-γ. The proteasome regulator PA28 was also expressed in every cell line and could be up-regulated by IFN-γ. In contrast, expression of the LMP2/LMP7 and TAP1/TAP2 varied between the different carcinoma cell lines (Fig. 5) and broadly correlated with their recognition by HPV16 E629–38-specific CTL. The highest levels of expression were seen in MDA231 cells. High levels of expression were also seen in HPV16+ SiHa cervical carcinoma cells although these were not used in CTL assays as they were HLA-A2-negative. Low to intermediate levels of immunoproteasome and TAP expression in CaSki and MS751 (apart from LMP7) cells were detected, but all could be up-regulated by IFN-γ. In contrast (and analogous to HLA-A*0201 expression), low levels of immunoproteasome and TAP expression in C33A and C33A-HPV16 cells were not inducible by IFN-γ (Fig. 5).
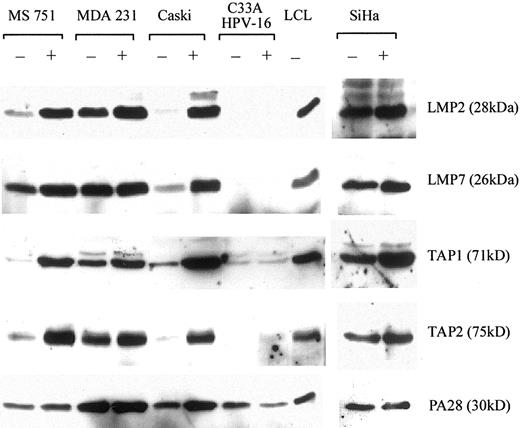
Cervical carcinoma cell lines express low levels of immunoproteasomes and TAP proteins that are up-regulated by IFN-γ. Carcinoma cell lines cultured with (+) or without (−) 200 U/ml of IFN-γ for 48 h were analyzed for expression of the immunoproteasome proteins LMP2 (28 kDa) and LMP7 (26 kDa), the transporter proteins TAP1 (71 kDa) and TAP2 (75 kDa), and the proteasomal regulator PA28 (30 kDa). The MUTU III B-LCL is included as a control.
Cervical carcinoma cell lines express low levels of immunoproteasomes and TAP proteins that are up-regulated by IFN-γ. Carcinoma cell lines cultured with (+) or without (−) 200 U/ml of IFN-γ for 48 h were analyzed for expression of the immunoproteasome proteins LMP2 (28 kDa) and LMP7 (26 kDa), the transporter proteins TAP1 (71 kDa) and TAP2 (75 kDa), and the proteasomal regulator PA28 (30 kDa). The MUTU III B-LCL is included as a control.
HPV16 E629–38-specific CTL recognize cervical carcinoma cells in the presence of IFN-γ and high levels of endogenous HPV16 E6 protein
IFN-γ was able to up-regulate the expression of multiple components of the class I processing pathway in most of the cervical carcinoma cell lines tested. We tested the effect of IFN-γ on the presentation of the HPV16 E629–38 by HPV16 cervical carcinomas CaSki and C33A-HPV16 (Fig. 6, A and F). Pretreatment of CaSki cells with IFN-γ resulted in a small (5%) increase in specific lysis by CTL, compared with untreated CaSki cells, but this was not considered significant (Fig. 6,A). There was no increase in the specific lysis of C33A-HPV16 cells treated with IFN-γ (Fig. 6 D).

HPV16 E629–38 peptide-specific CTL recognize CaSki cells in the presence of IFN-γ and high levels of endogenous HPV16 E6 protein. A, Clone 7E7 was assayed against 51Cr-labeled CaSki cells that had been cultured with (▪) or without (□) 200 U/ml IFN-γ for 48 h. The assay was repeated twice for both 3C11 and 7E7 with similar results. B, Clone 3C11 was assayed against 51Cr-labeled CaSki cells that were uninfected (□), infected with TA-HPV (•), or cultured with IFN-γ and then infected with TA-HPV (▴). The assay was repeated twice for both 3C11 and 7E7 with similar results. C, Clone 7E7 was assayed against 51Cr-labeled MS751 cells or (D) C33A-HPV16 cells that were either uninfected (□), infected with TA-HPV (•), or cultured with IFN-γ and then infected with TA-HPV (▴).
HPV16 E629–38 peptide-specific CTL recognize CaSki cells in the presence of IFN-γ and high levels of endogenous HPV16 E6 protein. A, Clone 7E7 was assayed against 51Cr-labeled CaSki cells that had been cultured with (▪) or without (□) 200 U/ml IFN-γ for 48 h. The assay was repeated twice for both 3C11 and 7E7 with similar results. B, Clone 3C11 was assayed against 51Cr-labeled CaSki cells that were uninfected (□), infected with TA-HPV (•), or cultured with IFN-γ and then infected with TA-HPV (▴). The assay was repeated twice for both 3C11 and 7E7 with similar results. C, Clone 7E7 was assayed against 51Cr-labeled MS751 cells or (D) C33A-HPV16 cells that were either uninfected (□), infected with TA-HPV (•), or cultured with IFN-γ and then infected with TA-HPV (▴).
Despite up-regulation of Ag-processing components by IFN-γ, there was no major effect on the presentation of HPV16 E629–38 by CaSki cells. This could result from an insufficient level of endogenous E6 expression or an inability of E6 to access the class I processing pathway. Consistent with this proposal was the finding that introduction of excess E6 protein by infection with TA-HPV (Fig. 6,B) allowed presentation of the HPV16 E629–38 epitope in IFN-γ-treated cells. Similarly, CTL recognition of IFN-γ-treated MS751 cell line was achieved by infecting with TA-HPV (Fig. 6,C). In contrast, C33A-HPV16 cells were not recognized by HPV16 E629–38 peptide-specific CTL even after combined treatment with IFN-γ and infection with TA-HPV (Fig. 6 D).
Presentation of the HPV16 E629–38 epitope is dependent on TAP
Intracellular processing of the HPV16 E629–38 peptide was investigated further using the HLA-A*0201 Ag-processing mutant cell line, LCL721.174 (.174) and transfectants derived from this cell line, .174/TAPs and .174TAPs/LMP7 (25). All three cell lines were recognized by HPV16 E629–38 peptide-specific CTL after pulsing with exogenous peptide (Fig. 7, A–C). The .174 cells lack the genes for TAP and LMP2/7 and, as expected, failed to present HPV16 E629–38 after TA-HPV infection (Fig. 7,A). In contrast, both .174/TAPs (Fig. 7,B) and .174/TAPs/LMP7 (Fig. 7,C) transfectants were able to present the HPV16 E629–38 epitope after TA-HPV infection. No improvement in recognition was seen by the additional transfection of the LMP7 gene (Fig. 7,C) into .174/TAPs (Fig. 7 B). These results suggest that HPV16 E629–38 is a TAP-dependent epitope and that LMP7 does not play a major role in generating the epitope.
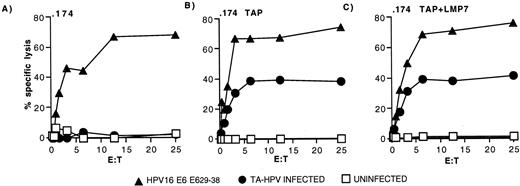
Presentation of the HPV16 E629–38 epitope is dependent on TAP. Clone 7E7 was assayed against 51Cr-labeled 721.174 cells (.174) (A), 721.174 cells transfected with the TAP1 and TAP2 genes (.174/TAPs) (B), and 721.174 cells transfected with the LMP7 gene as well as the TAP1 and TAP2 genes (.174/TAPs/LMP7) (C). Target cells were uninfected (□), infected with TA-HPV (•), or pulsed with HPV16 E629–38 (▴).
Presentation of the HPV16 E629–38 epitope is dependent on TAP. Clone 7E7 was assayed against 51Cr-labeled 721.174 cells (.174) (A), 721.174 cells transfected with the TAP1 and TAP2 genes (.174/TAPs) (B), and 721.174 cells transfected with the LMP7 gene as well as the TAP1 and TAP2 genes (.174/TAPs/LMP7) (C). Target cells were uninfected (□), infected with TA-HPV (•), or pulsed with HPV16 E629–38 (▴).
Discussion
Virtually all cervical cancers express the HPV oncogenes E6 and E7, thus providing attractive nonself-targets for CTL-based immunotherapy. This prospect has been tempered by several immunohistochemical studies demonstrating down-regulation or loss of HLA class I and TAP-1 molecules in cervical cancer lesions (46, 47, 48). However, these findings have not been related to functional assays. In this study we show for the first time that cervical carcinoma cell lines can have Ag-processing defects that limit the presentation of a peptide epitope derived from HPV16 E6.
We investigated Ag processing in cervical carcinoma cell lines using two HLA-A*0201-restricted CTL clones specific for HPV16 E629–38. Both CTL clones had similar specificity in that both could kill B-LCL expressing endogenous HPV E6 proteins and both had an avidity for peptide (50% maximal lysis at ∼1 nM) comparable to other tumor-specific CTL (49). Previous studies have suggested that the HPV16 E629–38 epitope is not a strong candidate for a CTL epitope as it only has intermediate binding affinity to HLA-A*0201, and has shown to be poorly immunogenic in vitro to human T cells (15). However, we have now demonstrated that this epitope can be generated after endogenous processing of HPV16 E6 protein (as supplied by recombinant vaccinia virus infection) in B-LCL and some epithelial cell lines. We also show for the first time that CTL against this epitope can be detected in patients with cervical carcinoma. How this CTL response is generated given the findings of the present study is unclear. In the recombinant adenovirus/DC restimulation system used to generate clone 3C11, we confirmed intracellular expression of HPV proteins in DCs. However, because the DC preparations were not 100% pure, and adenoviruses can infect a variety of cell types, we cannot exclude the possibility of indirect presentation of HPV Ags. Indeed, such a cross-priming mechanism might explain the low frequency of HPV16 E629–38-specific CTL responses detected in patients with cervical cancer (M. Evans, unpublished observations).
A striking finding was the inability of three cervical carcinoma cell lines (CaSki, C33A-HPV16, and MS751) to process and present the HPV16 E629–38 epitope to CTL clones even when excess HPV16 E6 protein was provided by recombinant vaccinia virus infection. This was not confined to HPV Ags as CaSki cells were also unable to process and present the HLA-A*0201-restricted influenza M158–66 epitope (our unpublished observations), and C33A cells fail to present certain EBV-derived epitopes (L.S. Young, personal communication). This defect correlated with the low expression of molecules involved in the Ag processing and presentation, such as HLA-A*0201, the immunoproteasome subunits LMP2 and LMP7, and TAP1 and TAP2. By contrast, cells with a high expression of these molecules such as the B-LCL C1R-A2 or the breast carcinoma MDA231 were capable of presenting HPV16 E629–38.
The phenotypes of the majority of the cervical and noncervical carcinoma cell lines tested were similar in that IFN-γ could up-regulate expression of Ag-processing components, as seen for other human cancers (50). The constitutive proteasome subunits were highly expressed by all the carcinoma cell lines tested and PA28 expressed at levels similar to B-LCL. C33A-HPV16 cells were distinct from the other carcinomas tested in that they had very low HLA-A*0201 expression and did not respond to IFN-γ. We were unable to detect LMP2 or LMP7 in HPV16-C33A cells, and TAP1 and TAP2 were barely detectable. This “severe” phenotype is similar to Ag-processing mutant B cell lines such as 721.174 or its derivative T2; however, incubation of HPV16-C33A with exogenous peptide or at low temperatures did not up-regulate HLA-A*0201 expression (our unpublished observations). These results suggest that in C33A cells, low expression of HLA-A*0201 is not controlled by the same mechanism as in T2 and that expression could be regulated at the transcriptional level.
Using 721.174 mutants, we found that presentation of the HPV16 E629–38 epitope was highly dependent on the presence of TAP. The processing and presentation of the HPV16 E629–38 epitope was found not to be overtly dependent on immunoproteasome subunit expression; normal expression of LMP7 (but low expression of TAP1, TAP2, and LMP2) in MS751 cells had no discernible effect, nor did transfection of LMP7 in 721.174 cells. The TAP dependence of the HPV16 E629–38 epitope and the requirement for IFN-γ treatment of vaccinia-infected Caski, and MS751 cells for CTL killing, suggest that the low levels of TAP in cervical carcinomas may be a limiting factor. However, our current data do not exclude a role for other IFN-γ-inducible components of the Ag-processing machinery in cervical carcinomas, including tapasin, chaperonins, and proteases (51). Direct proof that TAP is limiting in cervical carcinomas will require transfection of TAP1 and TAP2 genes; however, initial attempts involving double vaccinia infection (vaccinia-TAP plus vaccinia-HPV) have been unsuccessful (our unpublished observations).
Two of the three cervical carcinomas tested expressed HPV16 E6 protein either as a result of natural transformation (CaSki) or transfection (C33A-HPV16). However, despite the IFN-γ up-regulation of multiple components of the Ag-processing machinery in CaSki cells, these were still unable to present the HPV16 E629–38 epitope. Presentation was only seen when IFN-γ treatment was combined with expression of additional E6 protein following recombinant vaccinia virus infection. This suggests an inability to generate sufficient E629–38 from the endogenous source of E6 for CTL recognition. This may reflect constraints at the level of E6 expression or the efficiency with which E6 is processed and presented. CaSki cells contain between 60 and 600 copies of the HPV16 genome integrated at various sites. However, it has been difficult to accurately quantitate the levels of E6 protein in these cells (36). Immunoprecipitation studies have indicated that E7 is more abundant than E6 in cell lines (including CaSki) transformed by HPV16 and HPV18 (52, 53). It is possible that Ab detection of E6 protein is hindered by binding of other host cell proteins; this may also restrict the amounts of E6 accessible to the class I processing pathway. The major cellular target of E6 is p53, which is targeted for ubiquitin-mediated degradation via an intermediary E6-E6AP (E6 associated protein) complex. E6 is also capable of blocking the nuclear localization of p53 in response to DNA damage, by sequestering p53 in the cytoplasm, even in the absence of p53 degradation (54). Recently additional host cell proteins have been shown to bind to E6 (reviewed in Ref. 54). All of these factors could act to reduce the availability of E6 for degradation.
Interestingly, CaSki has been shown to carry an HPV16 variant that differs from the reference HPV16 sequence, in our vaccinia virus recombinants and HPV16-C33A, at two nucleotide positions in the E6 gene (55). Neither change is within the coding sequence for the E629–38 epitope, but these changes do result in nonconservative arginine to glycine, and leucine to tryptophan changes, 11 amino acids upstream and 53 amino acids downstream, respectively. Therefore, it cannot be excluded that these changes could exert an indirect effect on the processing of the epitope. Regardless of the mechanisms limiting the source of HPV E6 proteins naturally present in cervical carcinomas, it is clear that the Ag-processing defects are sufficient to limit the presentation of high levels of HPV16 E6 proteins introduced by recombinant vaccinia virus infection.
Our findings were based on the study of cervical carcinoma cell lines. This was largely done for technical reasons due to difficulties in obtaining patient biopsy material and the low success rate of establishing successful cell lines (56). We showed that the levels of TAP1, TAP2, LMP2, and LMP7 were low in three of the cervical carcinoma cell lines studied and were sufficient to limit presentation of an HPV-derived epitope to CTL. Although we cannot exclude changes resulting from extended in vitro culture, the situation in vivo is likely to be heterogeneous, and in some cases the phenotypes more severe. For example, immunohistochemical studies have shown complete loss of at least one HLA class I allele in up to 71% of cervical cancers (47), and coordinate loss of TAP1 and HLA class I in 36% of cervical carcinomas (46). By contrast, other studies have detected TAP in cervical carcinoma cell lines (albeit at lower levels than B-LCL), which could be up-regulated by IFN-γ (56). Similar findings have been reported in a immunohistochemical study of a small number of cervical carcinomas; TAP and LMP2 could be detected despite a loss of HLA class I in 28% of samples (57). All the above studies were performed with primary cervical carcinomas, but it is likely that the frequency of such defects may increase in cervical metastases such as those found in lymph nodes (48). In accordance with this the CaSki and MS751 cells used in the current study were derived from cervical tumor metases, whereas SiHA cells (HPV16+, normal expression of TAP1, TAP2 and LMP2, LMP7) were derived from a primary cervical tumor. Thus the cervical carcinoma cell lines studied represent the spectrum of cervical carcinoma phenotypes found in vivo.
The results from this study have several implications for the immunotherapy of cervical and other cancers. We have shown that some cervical carcinoma cell lines fail to present a TAP-dependent HPV16 E6 epitope. This appears to operate on at least two levels. First the source tumor protein E6 may be limiting, either due to the level of the protein expressed, or its ability to access the class I processing pathway. Second, the low levels of TAP and possibly other proteins involved in Ag processing in cervical carcinoma cell lines also act to limit processing even in the face of excess tumor E6 Ag. Immunohistochemical studies suggest that such defects do exist in human cancer tissue and may present a major obstacle to immunotherapy. However, such obstacles are not insurmountable. Several studies have shown that HPV-specific CTL directed against HLA-A*0201-restricted HPV16 E7 epitopes can kill CaSki cells (17, 19, 58) or C33A-HPV16 transfectants (27, 58). Our current findings would suggest that these epitopes are TAP-independent; however, this would have to be confirmed by testing appropriate CTL on vaccinia-HPV-infected .174 and .174-TAP transfectants as in Fig. 7. These epitopes may be atypical of HPV epitopes in general, as HLA-A*0201 has a strong binding affinity for TAP-independent peptides (59). Nevertheless, our findings suggest that future vaccine design should be biased toward similar CTL epitopes but restricted by multiple HLA class I molecules. This multiepitope approach combined with assessment of HLA class I molecules in tumor biopsies may allow individual tailoring of immunotherapy for cervical carcinoma.
We, and others (60), have used DCs expressing whole HPV Ags to generate HPV-specific CTL. However, the high levels of HLA class I, TAP, and immunoproteasomes expressed in DCs may select for CTL epitopes not presented by carcinoma cells. Therefore, novel methods such as those based on analysis of proteasomal digestion products (61), may be required to generate the multitude of CTL epitopes required to overcome Ag-processing defects in cervical carcinomas.
Acknowledgements
We thank Professor J. Dillner and Dr. L. Sherman for providing the C33A-HPV16 and MDA231 cell lines, respectively, Cantab Pharmaceuticals for providing TA-HPV, Wyeth, SR16, and SR18 vaccinia viruses, and Rod Dunbar for helpful advice on CTL cloning and expansion protocols. We are also grateful to Dr. R. Offringa and Dr. S. van der Burg for protocols and advice on the use of recombinant adenoviruses.
Footnotes
M.E. was supported by the Medical Research Council (U.K.); S.M., United Kingdom, U.G., and V.C. were supported by the Cancer Research Campaign. S.M. is a University Research Fellow of the Royal Society; M.R. and M.J. were supported by the Wellcome Trust.
Abbreviations used in this paper: HPV, human papillomavirus; B-LCL, B lymphoblastoid cell line; DC, dendritic cell; LMP, low molecular mass protein.
