

Abstract
Pseudomonas aeruginosa persists in patients with cystic fibrosis (CF) and drives CF lung disease progression. P. aeruginosa potently activates the innate immune system, mainly mediated through pathogen-associated molecular patterns, such as flagellin. However, the host is unable to eradicate this flagellated bacterium efficiently. The underlying immunological mechanisms are incompletely understood. Myeloid-derived suppressor cells (MDSCs) are innate immune cells generated in cancer and proinflammatory microenvironments and are capable of suppressing T cell responses. We hypothesized that P. aeruginosa induces MDSCs to escape T cell immunity. In this article, we demonstrate that granulocytic MDSCs accumulate in CF patients chronically infected with P. aeruginosa and correlate with CF lung disease activity. Flagellated P. aeruginosa culture supernatants induced the generation of MDSCs, an effect that was 1) dose-dependently mimicked by purified flagellin protein, 2) significantly reduced using flagellin-deficient P. aeruginosa bacteria, and 3) corresponded to TLR5 expression on MDSCs in vitro and in vivo. Both purified flagellin and flagellated P. aeruginosa induced an MDSC phenotype distinct from that of the previously described MDSC-inducing cytokine GM-CSF, characterized by an upregulation of the chemokine receptor CXCR4 on the surface of MDSCs. Functionally, P. aeruginosa–infected CF patient ex vivo–isolated as well as flagellin or P. aeruginosa in vitro–generated MDSCs efficiently suppressed polyclonal T cell proliferation in a dose-dependent manner and modulated Th17 responses. These studies demonstrate that flagellin induces the generation of MDSCs and suggest that P. aeruginosa uses this mechanism to undermine T cell–mediated host defense in CF and other P. aeruginosa–associated chronic lung diseases.
Introduction
Pseudomonas aeruginosa, a gram-negative flagellated bacterium, acts as opportunistic pathogen in immunocompromised hosts or compartments in which the local host defense is impaired. P. aeruginosa potently activates the innate arm of the immune system, an effect mainly mediated through pathogen-associated molecular patterns (PAMPs) and pattern recognition receptors. Among those, the TLR 5 ligand flagellin was found to play a key role in the recognition of P. aeruginosa (1–5). However, patients with chronic lung diseases, prototypically cystic fibrosis (CF) patients (6), are unable to eradicate the bacterium efficiently. The underlying immunological mechanisms are poorly understood.
P. aeruginosa is known to suppress T cell responses in vivo, and lymphocytes isolated from P. aeruginosa–infected CF patients show a blunted T cell proliferation capability ex vivo (7, 8). Beyond direct effects of P. aeruginosa on T cells (7, 8), this flagellated bacterium activates TLR5 on innate myeloid cells. Myeloid-derived suppressor cells (MDSCs) represent a novel innate immune cell subset generated in tumor, infective, and proinflammatory microenvironments (9, 10). These specialized innate immune cells are characterized by their capacity to suppress T cell responses and thereby modulate the cellular arm of adaptive immunity (10). Consequently, MDSCs are considered a key intermediary in balancing innate and adaptive immune responses, particularly under chronic disease conditions. In mice, in which MDSCs have been studied extensively, these cells constitute both a neutrophilic and a monocytic MDSC subphenotype (10–12); studies on MDSCs in human disease conditions are scarce. Recent evidence suggests that neutrophilic MDSCs accumulate in malignancy and systemic inflammation and are capable of suppressing T cell responses (13–15), but their role in host–pathogen interactions is, so far, poorly understood.
On the basis of the fact that CF patients are unable to eradicate P. aeruginosa infections and show impaired T cell proliferation, we hypothesized that P. aeruginosa induces MDSCs to escape T cell immunity. Our studies demonstrate that neutrophilic MDSCs are induced in P. aeruginosa–infected CF patients and correlate with pulmonary disease. Flagellated P. aeruginosa culture supernatants as well as purified flagellin dose-dependently and efficiently induced MDSC generation, corresponding to TLR5 expression on MDSCs. Functionally, both CF patient–isolated and flagellin-induced MDSCs suppressed T cell proliferation and modulated Th17 cells, as key antibacterial T cell populations in CF. This study highlights a novel mechanism by which the flagellated bacterium P. aeruginosa subverts host defense by inducing T cell–suppressive MDSCs. Given the well-documented role of this cell type in immune regulation, MDSCs could therefore represent a novel therapeutic target in CF and other diseases characterized by infections with flagellated bacteria.
Materials and Methods
Study subjects
The study was conducted at the University Children’s Hospital Tübingen (Tübingen, Germany). MDSCs were analyzed in the peripheral blood of patients with CF (n = 75) and age-matched pediatric non-CF control subjects without infections, malignancies, or inflammation (n = 37) (Table I). Informed written consent was obtained from all subjects included in the study or their legal representatives, and all study methods were approved by the local ethics committee. Inclusion criteria were the diagnosis of CF by clinical symptoms and positive sweat tests (sweat Cl− concentration > 60 mmol/l) or disease-causing mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Chronic P. aeruginosa infection was diagnosed if the organism was isolated in at least two consecutive sputum samples with a minimum interval of 6 mo. For lung function analyses, forced expiratory volume in 1 s (FEV1) and maximum expiratory flow rate at 25% of vital capacity (MEF25) were acquired and calculated. For the non–lung disease control group, samples were taken from children before they underwent elective surgery for multiple diagnoses. At the time of blood sampling, all control subjects were without any signs of infection, inflammation, or respiratory symptoms.
| Parameters | Cystic Fibrosis | Controls |
|---|---|---|
| N | 75 | 37 |
| Age (y) | 16 ± 10 | 14 ± 12 |
| Sex (m/f) | 38/37 | 21/16 |
| WBC (109/l) | 8 ± 2 | ND |
| CRP (mg/dl) | 0.41 ± 0.76 | ND |
| FVC (% pred.) | 98 ± 18 | ND. |
| FEV1 (% pred.) | 89 ± 22 | ND |
| MEF25 (% pred.) | 56 ± 30 | ND |
| P. aeruginosaa | 29 | ND |
| S. aureusb | 49 | ND |
| P. aeruginosa/S. aureus coinfection | 15 | |
| Age P. aeruginosa | 22 ± 11 | |
| Age S. aureus | 16 ± 9 | |
| Age P. aeruginosa/S. aureus coinfection | 21 ± 11 | |
| Antibiotics inhaled | 35 | ND |
| Antibiotics systemic | 14 | ND |
| dF508homozygous/heterozygous/other | 29/31/15 | ND |
| Parameters | Cystic Fibrosis | Controls |
|---|---|---|
| N | 75 | 37 |
| Age (y) | 16 ± 10 | 14 ± 12 |
| Sex (m/f) | 38/37 | 21/16 |
| WBC (109/l) | 8 ± 2 | ND |
| CRP (mg/dl) | 0.41 ± 0.76 | ND |
| FVC (% pred.) | 98 ± 18 | ND. |
| FEV1 (% pred.) | 89 ± 22 | ND |
| MEF25 (% pred.) | 56 ± 30 | ND |
| P. aeruginosaa | 29 | ND |
| S. aureusb | 49 | ND |
| P. aeruginosa/S. aureus coinfection | 15 | |
| Age P. aeruginosa | 22 ± 11 | |
| Age S. aureus | 16 ± 9 | |
| Age P. aeruginosa/S. aureus coinfection | 21 ± 11 | |
| Antibiotics inhaled | 35 | ND |
| Antibiotics systemic | 14 | ND |
| dF508homozygous/heterozygous/other | 29/31/15 | ND |
Results are expressed as means ± SD.
P. aeruginosa bacteria isolated in at least two consecutive sputum samples or throat swabs with a minimum interval of 6 mo.
S. aureus bacteria isolated in at least two consecutive sputum samples or throat swabs with a minimum interval of 6 mo.
% pred., % of predicted; CRP, C-reactive protein; f, female; FVC, forced vital capacity; m, male.
Cell isolation and flow cytometry
PBMCs were prepared from blood samples by Ficoll density gradient sedimentation (Lymphocyte Separation Medium; Biochrom) and washed twice in RPMI 1640 medium. Trypan blue staining solution at 0.5% differentiated between viable and nonviable cells and showed viability of >90% for all cells used in this study. After Ficoll density gradient sedimentation, MDSCs were characterized as CD33highCD66bhighIL-4RαinterHLA-DRdim neutrophilic cells in the PBMC fraction, according to previously established human MDSC analysis methods (13, 15) (Fig. 1). For MDSC isolation, cells were obtained from the PBMC fraction and labeled with anti–CD66b-FITC, followed by two sequential anti-FITC magnetic bead separation steps (Miltenyi Biotec), per the manufacturer’s protocol. Purity of CD66b+ cells after separation was >95%, as assessed by flow cytometry. Morphology of the cells (MDSCs and conventional neutrophils isolated from healthy control individuals) was performed by staining of cytospins. Isolated MDSCs showed typical morphological characteristics of neutrophils, marking them as neutrophilic MDSCs (see supplementary material). For cytospin stainings, 5 × 104 cells were centrifuged in a Cytospin 3 Centrifuge (Shandon) at 800 rpm for 15 min and stained after with May–Grünwald–Giemsa. Abs against CD3, CD4, CD8, CD14, CD16, CD66b, HLA-DR, and CD124 (IL-4Rα) were purchased from BD Pharmingen. Abs against CD11b and CD33 were purchased from Miltenyi Biotec. Anti-human CXCR4 (clone 12G5) was obtained from eBioscience (San Diego, CA). Abs against TLR5 were purchased from Santa Cruz Biotechnology. Mouse IgG1-FITC, Mouse IgM-FITC, Mouse IgG1-PE, and Mouse IgG1-APC (BD Pharmingen) were used as isotype controls. Results were expressed as percent of positive cells and mean fluorescence intensity. Calculations were performed with BD CellQuest analysis software. For Th17 cell staining, 2.5 × 106 PBMCs were stimulated overnight with 10 ng/ml PMA and 1 μg/ml ionomycin (Sigma-Aldrich, St Louis, MO) in the presence of GolgiPlug (BD Biosciences, San Jose, CA). After cell surface staining with PE-conjugated anti-CD4 (eBioscience), cells were fixed and permeabilized (Cytofix/Cytoperm; BD Biosciences) and stained with Alexa Fluor 647–conjugated anti–IL-17A (eBioscience). As a control for cellular activation and intracellular staining, CD4+ T cells were also evaluated for IFN-γ production (FITC–conjugated; eBioscience). For all in vitro FACS assays, three independent experiments were performed.
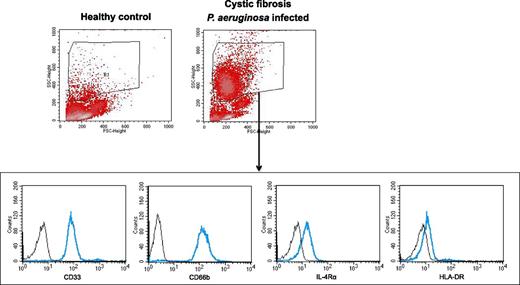
Characterization of human MDSCs. MDSCs were characterized in the forward scatter (FSC)/side scatter (SSC) area as a granulocytic cell population in the PBMC fraction (low-density neutrophils) (R1), as published previously (13, 15). This population was distinct from lymphocytes, monocytes, erythrocytes, or debris. Within R1, neutrophilic MDSCs were identified as a CD33highCD66bhighIL-4RαinterHLA-DRdim–expressing neutrophilic MDSC population. MDSCs are shown in a representative healthy control subject and a CF patient infected with P. aeruginosa.
Characterization of human MDSCs. MDSCs were characterized in the forward scatter (FSC)/side scatter (SSC) area as a granulocytic cell population in the PBMC fraction (low-density neutrophils) (R1), as published previously (13, 15). This population was distinct from lymphocytes, monocytes, erythrocytes, or debris. Within R1, neutrophilic MDSCs were identified as a CD33highCD66bhighIL-4RαinterHLA-DRdim–expressing neutrophilic MDSC population. MDSCs are shown in a representative healthy control subject and a CF patient infected with P. aeruginosa.
In vitro generation and isolation of human MDSCs
Human MDSCs were generated in vitro according to a previously published protocol (16). Isolated human PBMCs were cultured in 12-well flat-bottom plates (Corning) or 25-cm2 flasks (Greiner Bio-One) at 5 × 105 cells/ml in complete medium for 6 d, and GM-CSF (10 ng/ml; Genzyme), P. aeruginosa culture supernatants (0.1–1%), flagellin (0.0001–0.1 μg/ml; Invivogen), or the CFTR inhibitors CFTRinh-172 (10 μM; Tocris) or GlyH-101 (10 μM; Merck, Millipore) were added as indicated in the respective figures. For all assays, at least three independent experiments were performed. P. aeruginosa culture supernatants were generated from two flagellated (PAO1 and PA14) strains or from a flagellin-deficient (PAO1 fliM) strain according to standard protocols. PBMCs cultured in medium alone were run in parallel as a control for each donor. Medium and supplements were refreshed after 3 d. After 6 d, all cells were collected from PBMC cultures. Adherent cells were removed using the nonprotease cell detachment solution Detachin (Genlantis). CD33+ cells were isolated from each culture, using anti-CD33 magnetic microbeads and LS column separation (Miltenyi Biotec) according to the manufacturer’s instructions. The purity of isolated cell populations was >90% by flow cytometry.
T cell suppression assay
Responder PBMCs were obtained from healthy volunteers and stained with CFSE according to the manufacturer's protocol (Invitrogen). PBMCs were stimulated with 100 U/ml IL-2 (R&D Systems) and 1 μg/ml OKT3 (Janssen-Cilag). In a standardized way, 60,000 PBMCs per well in RPMI 1640 (Biochrom) were seeded in a 96-well microtiter plate, and 10,000–30,000 MDSCs in RPMI 1640 or, as control, isolated conventional non-MDSC neutrophils in RPMI 1640 or RPMI1640 only was added. The cell culture was supplemented with 10% heat-inactivated human serum, 2 mM glutamine, 100 IU/ml penicillin, and 100 mg/ml streptomycin. After 96 h of incubation in a humidified atmosphere at 37°C and 5% CO2, cells were harvested and supernatants were frozen in −20°C. CFSE-fluorescence intensity was analyzed by flow cytometry to determine polyclonal T cell proliferation. For all assays, at least three independent experiments were performed. CD4 or CD8 T cells, respectively, were gated out for CFSE dilution after polyclonal stimulation. Only propidium iodide–negative cells were considered for analysis. Where indicated (Supplemental Fig. 3), dose-dependent MDSC–T cell experiments were performed. Where indicated, the effect of CXCR4 inhibition, using the small-molecule inhibitor AMD3100 (2 μM; Sigma-Aldrich), on MDSC-mediated suppression of CD4 and CD8 T cell proliferation was analyzed (Supplemental Fig. 2C).
Quantitative PCR
Quantitative RT-PCR was performed in MACS-isolated human MDSCs. mRNA was isolated with the NucleoSpin RNA II kit (Macherey & Nagel, Düren, Germany), cDNA was synthesized with the iScript Advanced Synthesis Kit (Bio-Rad, Munich, Germany), and real-time RT-PCR was performed using the Power SYBR Green Master Mix (Applied Biosystems, Darmstadt, Germany) and a ViiA7 Real-Time PCR cycler (Applied Biosystems) according to the manufacturer’s protocols. Results were calculated by the 2(−ΔΔCT) method and are given as relative expression in comparison with the housekeeping gene β-actin. The following primers were used:
G-CSF, forward: 5′-TAGAGCAAGTGAGGAAGATCAGG-3′,
reverse: 5′-AGTTCTTCCATCTGCTGCCAGATG-3′;
β-actin: forward: 5′-CATGTACGTTGCTATCCAGGC-3′,
reverse: 5′-CTCCTTAATGTCACGCACGAT-3′.
Cytokine analysis in culture supernatants
Multiplex cytokine array analysis in supernatants was performed using the Bio-Plex protein multiarray system (Bio-Plex Pro Human Cytokine Array, Bio-Rad Laboratories). For the current experiments, a human 27-plex assay was used according to the recommendations of the manufacturer (Bio-Rad).
Statistical analysis
Statistical analysis was accomplished using GraphPad Prism 5.0 (GraphPad Software). Differences between the groups were determined by a Student t test. A p value < 0.05 was considered significant.
Results
Neutrophilic MDSCs accumulate in Pseudomonas aeruginosa–infected CF patients
We studied patients with CF lung disease, characterized by P. aeruginosa infections. In a large cross-sectional CF patient cohort, percentages of MDSCs (Fig. 1) were significantly increased compared with percentages in age-matched healthy control subjects (Fig. 2A). This accumulation of MDSCs was not due to relative changes in peripheral blood mononuclear, polymorphonuclear cell counts, or band cells. Scatter plot analyses further revealed a high interpatient variability, ranging from MDSC percentages in CF patients similar to those in control individuals (<1% of PBMCs) to CF patients with up to 20% MDSCs (of PBMCs gated), suggesting underlying disease-associated factors regulating MDSC generation in CF patients.
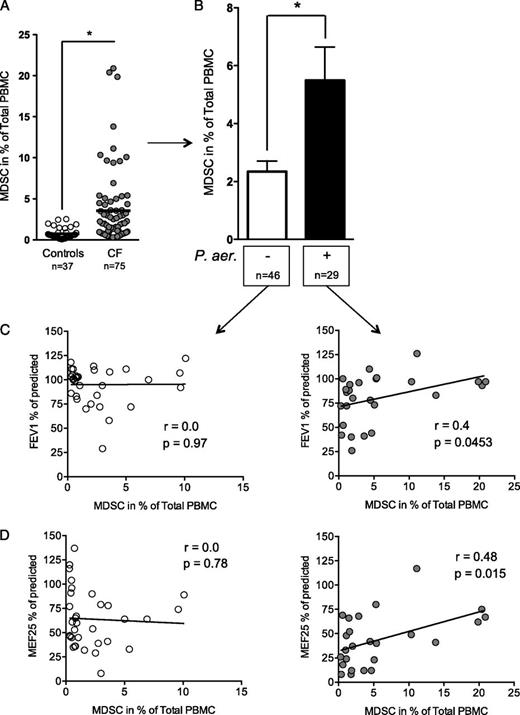
Increased MDSCs in chronic P. aeruginosa infection. (A) Percentages of MDSCs in healthy control subjects (white circles) and in CF patients (gray circles). (B) Percentages of MDSCs in non–P. aeruginosa-infected (white bar) and P. aeruginosa–infected (black bar) CF patients. (C and D) Correlation between percentages of MDSCs and obstructive lung function parameters (FEV1 and MEF25) in non–P. aeruginosa-infected (left panel) and P. aeruginosa–infected (right panel) CF patients. *p < 0.05.
Increased MDSCs in chronic P. aeruginosa infection. (A) Percentages of MDSCs in healthy control subjects (white circles) and in CF patients (gray circles). (B) Percentages of MDSCs in non–P. aeruginosa-infected (white bar) and P. aeruginosa–infected (black bar) CF patients. (C and D) Correlation between percentages of MDSCs and obstructive lung function parameters (FEV1 and MEF25) in non–P. aeruginosa-infected (left panel) and P. aeruginosa–infected (right panel) CF patients. *p < 0.05.
Patient stratification showed that, in particular, CF patients chronically infected with P. aeruginosa showed significantly increased MDSC percentages over those in CF patients without P. aeruginosa infection (Figs. 1, 2B). The majority of P. aeruginosa–infected CF patients had a nonmucoid Pseudomonas phenotype (86%), and percentages of MDSCs tended to be higher in CF patients with nonmucoid than in patients with mucoid P. aeruginosa isolates (Supplemental Fig. 1A). Importantly, percentages of MDSCs correlated positively with obstructive pulmonary function parameters (FEV1: r = 0.4, p = 0.0045; MEF25: r = 0.48, p = 0.015) in chronically P. aeruginosa–infected, but not in non–Pseudomonas-infected CF patients (Fig. 2C, 2D). Longitudinal follow-up of individual CF patients indicated that percentages of MDSCs increased in P. aeruginosa–infected, but not in non–P. aeruginosa–infected, conditions, and also suggested that MDSCs correlate with lung function over time (Supplemental Fig. 1B). No statistical association of antibiotics with MDSCs was found. Despite a substantial number of CF patients coinfected with P. aeruginosa and Staphylococcus aureus (15 of 29, see Table I for details), no statistical associations were found between MDSC percentages and the detection of S. aureus or Aspergillus fumigatus, as other characteristic pathogens in CF lung disease, suggesting that the accumulation of MDSCs in CF patients is not due to infection in general but indicates that P. aeruginosa–associated factors induce MDSCs and thereby modulate lung disease severity in CF.
Flagellated Pseudomonas aeruginosa and purified flagellin induce CXCR4high MDSCs
To test our hypothesis that P. aeruginosa–derived factors, such as microbe-associated or shedded PAMPs (6), in CF patients drive the generation of MDSCs, we first used P. aeruginosa culture supernatants from two different P. aeruginosa strains and tested their capacity to induce MDSC generation in vitro. These studies demonstrated that P. aeruginosa culture supernatants harvested from two different flagellated P. aeruginosa strains efficiently induced MDSCs to a similar extent as GM-CSF did, which is well known to induce MDSCs in this setting in vitro (16) (Fig. 3A).
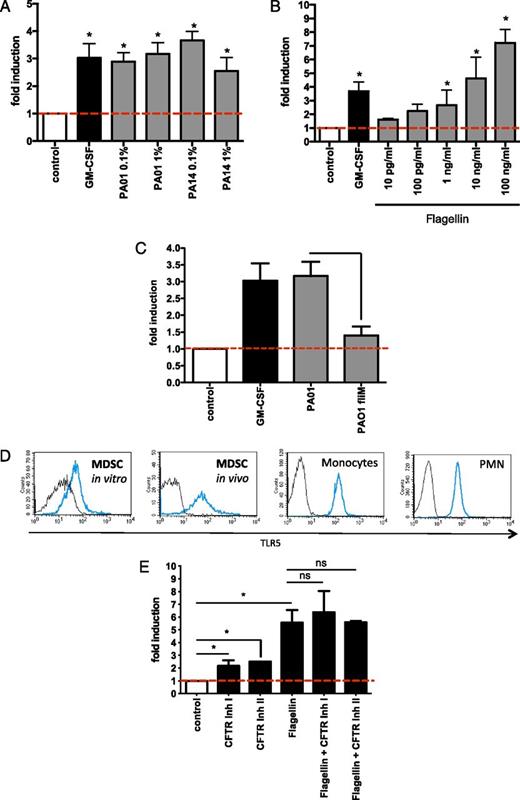
Flagellated P. aeruginosa bacteria and purified flagellin protein induce MDSCs. (A) Flagellated P. aeruginosa induces MDSCs. MDSCs were generated by incubating PBMCs with medium only (negative control), GM-CSF (10 ng/ml, positive control), or two different flagellated P. aeruginosa strain (PAO1 or PA14) derived culture supernatants at 0.1 or 1% medium culture conditions. The x-fold induction of MDSCs compared with control conditions is depicted. (B) Flagellin induces MDSCs. The effect of purified flagellin (for concentrations, see figure) or GM-CSF (10 ng/ml) on MDSC generation is depicted. The x-fold induction of MDSCs compared with control conditions is depicted. (C) P. aeruginosa induces MDSCs through flagellin. MDSCs were generated by incubating PBMCs with medium only (negative control), with GM-CSF (10 ng/ml, positive control) or with culture supernatants (1% medium culture conditions) from a flagellated P. aeruginosa strain (PAO1) or a matched nonflagellated strain (PAO1 fliM mutant strain). The x-fold induction of MDSCs compared with control conditions is depicted. (D) MDSCs express TLR5. MDSCs were generated in vitro by incubating isolated PBMCs with P. aeruginosa PAO1 (1%) derived culture supernatants (“MDSC in vitro”). TLR5 expression on CF patient ex vivo–isolated MDSCs (“MDSC ex vivo”). As control immune cells, peripheral blood monocytes and neutrophils (PMNs) were stained for TLR5. TLR5 surface expression (blue line) versus unstained control (black line) is shown. (E) MDSC induction and CFTR. MDSCs were generated in vitro by incubating isolated PBMCs with purified flagellin (10 ng/ml), with or without pretreatment of PBMCs with two different small-molecule CFTR inhibitors (CFTRinh-172, 10μM; GlyH-101, 10μM). For all assays, at least three independent experiments were performed. *p < 0.05. ns, Not significant.
Flagellated P. aeruginosa bacteria and purified flagellin protein induce MDSCs. (A) Flagellated P. aeruginosa induces MDSCs. MDSCs were generated by incubating PBMCs with medium only (negative control), GM-CSF (10 ng/ml, positive control), or two different flagellated P. aeruginosa strain (PAO1 or PA14) derived culture supernatants at 0.1 or 1% medium culture conditions. The x-fold induction of MDSCs compared with control conditions is depicted. (B) Flagellin induces MDSCs. The effect of purified flagellin (for concentrations, see figure) or GM-CSF (10 ng/ml) on MDSC generation is depicted. The x-fold induction of MDSCs compared with control conditions is depicted. (C) P. aeruginosa induces MDSCs through flagellin. MDSCs were generated by incubating PBMCs with medium only (negative control), with GM-CSF (10 ng/ml, positive control) or with culture supernatants (1% medium culture conditions) from a flagellated P. aeruginosa strain (PAO1) or a matched nonflagellated strain (PAO1 fliM mutant strain). The x-fold induction of MDSCs compared with control conditions is depicted. (D) MDSCs express TLR5. MDSCs were generated in vitro by incubating isolated PBMCs with P. aeruginosa PAO1 (1%) derived culture supernatants (“MDSC in vitro”). TLR5 expression on CF patient ex vivo–isolated MDSCs (“MDSC ex vivo”). As control immune cells, peripheral blood monocytes and neutrophils (PMNs) were stained for TLR5. TLR5 surface expression (blue line) versus unstained control (black line) is shown. (E) MDSC induction and CFTR. MDSCs were generated in vitro by incubating isolated PBMCs with purified flagellin (10 ng/ml), with or without pretreatment of PBMCs with two different small-molecule CFTR inhibitors (CFTRinh-172, 10μM; GlyH-101, 10μM). For all assays, at least three independent experiments were performed. *p < 0.05. ns, Not significant.
To dissect which mechanism(s) induce(s) MDSCs in chronic P. aeruginosa infection, we focused on flagellin because 1) infection with flagellated P. aeruginosa was associated with MDSC induction in our CF patient cohort, whereas nonflagellated microbes did not show any association with MDSCs in vivo; and 2) previous studies showed that among different PAMPs, flagellin recognition through TLR5 in particular plays a key role in leukocyte–P. aeruginosa interactions in CF lung disease (4, 5, 17, 18). Our studies demonstrated that flagellin efficiently and dose-dependently induced MDSCs with a more potent capacity than GM-CSF did (Fig. 3B). Further studies also showed that flagellin upregulated G-CSF, but had no effect on GM-CSF expression in MDSCs (Supplemental Fig. 2A and data not shown). P. aeruginosa–induced MDSC generation was significantly reduced using a flagellin-deficient P. aeruginosa strain (Fig. 3C). Because flagellin acts mainly through TLR5, we analyzed TLR5 surface expression on MDSCs and found that both in vitro–generated and P. aeruginosa CF patient in vivo/ex vivo isolated MDSCs expressed TLR5 (Fig. 3D). To assess the effect of the CFTR in MDSC generation, we used a small-molecule CFTR inhibitor. These studies demonstrated that CFTR inhibition slightly increased the generation of MDSCs but had no significant effect on flagellin-induced MDSC generation (Fig. 3E). These findings were not due to toxic or proapoptotic effects of the CFTR inhibitor and could be reproduced using a second different CFTR inhibitor (Fig. 3E).
Next, we characterized the immunological phenotype of P. aeruginosa– and flagellin-induced MDSCs compared with canonical GM-CSF–induced MDSCs. These studies demonstrated that both flagellated P. aeruginosa bacteria and purified flagellin protein upregulated surface expression levels of the homeostatic chemokine receptor and HIV coreceptor CXCR4 on MDSCs, whereas GM-CSF treatment had no significant effect on the expression of these surface proteins (Fig. 4). When viewed in combination, these studies indicate that flagellated P. aeruginosa strains and purified flagellin protein potently induce MDSCs with a distinct CXCR4high MDSC phenotype.

P. aeruginosa and flagellin upregulate CXCR4 expression on MDSCs. CXCR4 surface expression levels were quantified on GM-CSF, P. aeruginosa PAO1 (1%), or flagellin (10 ng/ml) in vitro–generated MDSCs. For all assays, three independent experiments were performed. Where indicated, the small-molecule CFTR inhibitor CFTRinh-172 (10 μM) was used. *p < 0.05.
P. aeruginosa and flagellin upregulate CXCR4 expression on MDSCs. CXCR4 surface expression levels were quantified on GM-CSF, P. aeruginosa PAO1 (1%), or flagellin (10 ng/ml) in vitro–generated MDSCs. For all assays, three independent experiments were performed. Where indicated, the small-molecule CFTR inhibitor CFTRinh-172 (10 μM) was used. *p < 0.05.
Flagellin-induced MDSCs functionally suppress T cell proliferation ex vivo and in vitro
To assess the functional capacities of flagellated P. aeruginosa–induced phenotypic MDSCs and to corroborate their role as a T cell suppressive cell type, we isolated MDSCs from P. aeruginosa–infected CF patients, using gradient centrifugation and sequential magnetic bead isolation, and studied MDSC–T cell interactions ex vivo. These studies using T cell CFSE labeling demonstrated that P. aeruginosa–infected CF patient–derived MDSCs efficiently suppressed polyclonal T cell proliferation of both CD4+ and CD8+ T cell subsets in a dose-dependent fashion (Fig. 5A, Supplemental Fig. 2B). As flagellin induced CXCR4high MDSCs, we investigated whether inhibiting CXCR4 functionality using the small-molecule inhibitor AMD3100 has an effect on MDSC-mediated T cell suppression. These studies showed that CXCR4 inhibition had no effect on MDSC-mediated T cell suppression (Supplemental Fig. 2C).

T cell suppression by CF MDSCs. (A) The suppressive effect of CD66b+–MACS-isolated CF MDSCs was analyzed on CD4+ and CD8+ T cell subsets, using the CFSE polyclonal proliferation assay. (B) The effect of CD66b+–MACS-isolated CF MDSCs on IL-17 secretion by CD4+ T cells (Th17 cells) was analyzed.
T cell suppression by CF MDSCs. (A) The suppressive effect of CD66b+–MACS-isolated CF MDSCs was analyzed on CD4+ and CD8+ T cell subsets, using the CFSE polyclonal proliferation assay. (B) The effect of CD66b+–MACS-isolated CF MDSCs on IL-17 secretion by CD4+ T cells (Th17 cells) was analyzed.
Because IL-17–producing T cells have been previously described as playing a pivotal role in the pathogenesis of chronic infective CF lung disease, we investigated whether CF-isolated MDSCs are capable of modulating IL-17 protein production by CD4+ T cells. These studies demonstrated that MDSCs derived from P. aeruginosa–infected CF patients substantially dampened released IL-17 protein (64% reduction of IL-17 protein) (Fig. 5B). These findings on IL-17 cytokine production, quantified in cell culture supernatants, were confirmed by intracellular flow cytometry stainings, showing that MDSCs suppressed IL-17 protein expression in T cells (Supplemental Fig. 2D). In addition to IL-17, MDSCs had an effect on several other cytokines, chemokines, and growth factors, as analyzed by a high-throughput bioplex array (Supplemental Fig. 3).
Finally, we investigated whether P. aeruginosa– or flagellin-induced MDSCs feature a similar T cell–suppressive phenotype. These studies showed that flagellated P. aeruginosa–or purified flagellin-induced MDSCs mimicked the T cell–suppressive characteristics of CF patient ex vivo–isolated MDSCs in potently suppressing both CD4 and CD8 T cell proliferation (Fig. 6). When viewed in combination, these studies demonstrate that both ex vivo CF patient–isolated and in vitro P. aeruginosa–/flagellin-induced MDSCs suppress CD4+ and CD8+ T cell proliferation and modulate Th17 responses.

T cell suppression by P. aeruginosa– or flagellin-induced MDSCs. The suppressive effect of MDSCs induced by P. aeruginosa PAO1 (1%) or flagellin (10 ng/ml) was analyzed on CD4+ and CD8+ T cell subsets, using the CFSE polyclonal proliferation assay.
T cell suppression by P. aeruginosa– or flagellin-induced MDSCs. The suppressive effect of MDSCs induced by P. aeruginosa PAO1 (1%) or flagellin (10 ng/ml) was analyzed on CD4+ and CD8+ T cell subsets, using the CFSE polyclonal proliferation assay.
Discussion
P. aeruginosa survives in immunocompromised individuals and patients with chronic pulmonary diseases, such as CF, owing to the inability of the host to clear this pathogen efficiently. However, the underlying immune mechanisms remain poorly understood (6). This study reveals a novel mechanism by which flagellated P. aeruginosa bacteria subvert the host defense by inducing T cell–suppressive MDSCs. MDSCs could therefore represent a novel therapeutic target in CF and other diseases characterized by infections with P. aeruginosa or other flagellated bacteria.
MDSCs represent an innate immune cell subset that efficiently controls adaptive T cell inflammation (9, 10). Our studies provide evidence that granulocytic/neutrophilic MDSCs, regularly absent or only marginally present in healthy control individuals, accumulate in P. aeruginosa–infected CF patients. We further show that flagellated P. aeruginosa bacteria or the purified TLR5 ligand flagellin potently induces T cell–suppressive MDSCs and upregulates CXCR4 on MDSCs. Importantly, flagellin-deficient P. aeruginosa bacteria were significantly impaired in inducing MDSCs, supporting the idea that the effect of P. aeruginosa in vivo and in vitro is mainly mediated through flagellin. The significance of the flagellin–TLR5 axis in P. aeruginosa–host interactions has been previously demonstrated in numerous studies, including experimental infection models using epithelial cells (1, 2, 5, 17, 19, 20), macrophages (21), neutrophils (18), or in vivo infection strategies (3, 22, 23). Our study thereby confirms and extends the concept that flagellin acts as a key player in shaping the innate but also the adaptive immune response to P. aeruginosa in CF lung disease and beyond.
On the basis of our findings, we speculate that flagellin-induced MDSC accumulation in chronic P. aeruginosa infection in CF patients has functional immunological relevance, as ex vivo and in vitro P. aeruginosa–induced MDSCs significantly suppressed both CD4 and CD8 T cell proliferation and dampened Th17 cell responses, which play a vital role in CF lung disease (24–31). Consequently, MDSCs, as key interface cells between innate and adaptive immunity, may represent a novel therapeutic target in CF and other P. aeruginosa–associated pulmonary diseases, such as chronic obstructive pulmonary disease or ventilator-associated pneumonia. Beyond these lung diseases and wound infections, in which P. aeruginosa also plays an important part, our findings may have an even broader immunological relevance for understanding host–pathogen interactions, because P. aeruginosa infection was previously found to cross-suppress T cell responses and immunity to other bacterial pathogens in vivo (8, 32). Although direct effects of P. aeruginosa toxins on T cells probably also have a function in this setting (8, 32), our finding that MDSCs directly isolated from P. aeruginosa–infected patients potently suppressed T cells ex vivo in the absence of P. aeruginosa products supports the notion that MDSCs represent a distinct mechanism mediating flagellated P. aeruginosa–induced immune suppression.
Previous studies have characterized the T cell response in human CF patients (33, 34) (24, 26, 27, 30) (35) and murine P. aeruginosa (36–40) or Aspergillus fumigatus infection models (41–43), providing evidence that the adaptive T cell response in CF is altered at several levels. CF T cells were found to be prone to a Th2 phenotype, with the CFTR mutation itself and/or chronic infections with P. aeruginosa as possible underlying factors (42, 44–46). Importantly, recent lines of evidence indicate that Th17 cells and the IL-17 axis–related cytokines IL-22 and IL-23 could play an essential role in recruiting neutrophils to the CF airways and thereby contributing to neutrophil-mediated tissue damage in CF lung disease, thus labeling CF as a “Th17 disease” (24–31). Th17 cells secrete IL-17 that, in turn, enhances G-CSF and thereby boosts neutrophil mobilization from the bone marrow. The open question remains how P. aeruginosa is able to colonize and chronically infect the CF airways in the face of this activated antibacterial Th17 response, suggesting mechanisms that modulate and impair Th17 immunity in CF.
Combining these views and our data on MDSCs into one unifying pathophysiological scenario, we propose the following regulatory loop: 1) P. aeruginosa induces MDSCs through a flagellin-mediated mechanism; 2) MDSCs dampen T cell proliferation and Th17 cell responses and thereby protect P. aeruginosa from T cell–mediated host defense; and 3) MDSCs downregulate neutrophil recruitment by inhibiting IL-17 release, thereby preventing tissue damage by the unbalanced release of neutrophil-derived proteases and oxidants (47). On the basis of our unexpected observation that an increase of MDSCs correlated with improved pulmonary function in P. aeruginosa–infected CF patients, we speculate that the MDSC-mediated downregulation of Th17 responses dampens neutrophilic lung tissue damage and could therefore beneficially modulate the course of CF lung disease, a conception necessitating future investigations. This “anti-inflammatory” action of MDSCs could be of major relevance for progressive stages of CF lung disease, when damage-associated molecular patterns, neutrophil-derived proteases, and protease-generated extracellular breakdown products, such as proline-glycine-proline, further sustain the neutrophil influx and perpetuate IL-17–driven inflammation (48, 49). As 1) P. aeruginosa–uninfected CF patients showed increased MDSCs compared with healthy controls, 2) CFTR inhibition had a slight effect on MDSC induction, and 3) flagellin induced host-derived G-CSF expression in vitro, the CFTR mutation itself and non–P. aeruginosa host-derived factors probably also contribute to MDSC induction in the course of CF lung disease.
This study demonstrates that flagellin potently induces MDSC generation and that this PAMP mediates the effects of the CF-associated pathogen P. aeruginosa on MDSC generation. In chronic CF lung disease, P. aeruginosa becomes mucoid, thereby preventing flagellin-host cell interactions. Because our CF patient cohort was characterized by a clear predominance of nonmucoid P. aeruginosa isolates and MDSCs tended to be higher in CF patients featuring nonmucoid than mucoid P. aeruginosa isolates, we speculate that P. aeruginosa–derived flagellin plays an essential role in MDSC induction, at least in pediatric CF cohorts. However, future studies in adult CF patients with a higher proportion of mucoid P. aeruginosa phenotypes, using methods such as the swimming motility assays (5), are required to assess whether motile P. aeruginosa and functional flagella are required to induce MDSCs in CF in vivo. Moreover, representative longitudinal studies should address the question whether MDSCs can be predictive for the course of CF lung disease, such as disease exacerbations.
When viewed in combination, this study demonstrates that P. aeruginosa infection induces MDSCs and subverts T cell immunity, mediated through a novel flagellin-dependent immune mechanism. MDSCs may therefore represent a hitherto unappreciated therapeutic target to modulate inflammation and host defense in CF and other diseases characterized by P. aeruginosa infection, such as chronic obstructive pulmonary disease, ventilator-associated pneumonia, wound infections, or primary ciliary dyskinesia, in which we also observed increased MDSCs (N. Rieber and D. Hartl, unpublished observations). Beyond P. aeruginosa, these findings may have a broader relevance for infections with flagellated bacteria in general, such as Helicobacter pylori, Salmonella typhimurium, or flagellated Escherichia coli strains. The flagellin-induced upregulation of the G-protein–coupled receptor CXCR4 on MDSCs could pave the way to target this leukocyte subset pharmacologically, using small-molecule inhibitors and underpins, with CXCR4 as HIV coreceptor, a potential role for MDSCs in HIV infection (50). Our observation that flagellin induced CXCR4high MDSCs, but that CXCR4 was dispensable for MDSC-mediated T cell suppression, may further suggest that CXCR4 in this setting may play a role in MDSC functionality beyond T cell suppression, such as MDSC migration, homing, or survival. Because signal transducer and activator of transcription 3 (STAT3), S100 proteins, and NO have been previously implicated in the generation and functionality of MDSCs (10), interfering with these pathways might represent a future therapeutic strategy in CF lung disease and other pathological conditions associated with flagellated bacteria
Footnotes
This work was supported by the German Research Foundation, Emmy Noether Programme HA 5274/3-1, the German Society of Pediatric Pneumonology, Präventions- und Informationsnetzwerk Allergie/Asthma, the Novartis Foundation, and the Ernest-Solvay-Foundation (all to D.H.).
The online version of this article contains supplemental material.
References
Disclosures
The authors have no financial conflicts of interest.
