

Abstract
The transcription factor NF-κB stimulates the transcription of proinflammatory cytokines including TNF-α. LPS (endotoxin) and hypoxia both induce NF-κB activation and TNF-α gene transcription. Furthermore, hypoxia augments LPS induction of TNF-α mRNA. Previous reports have indicated that antioxidants abolish NF-κB activation in response to LPS or hypoxia, which suggests that reactive oxygen species (ROS) are involved in NF-κB activation. This study tested whether mitochondrial ROS are required for both NF-κB activation and the increase in TNF-α mRNA levels during hypoxia and LPS. Our results indicate that hypoxia (1.5% O2) stimulates NF-κB and TNF-α gene transcription and increases ROS generation as measured by the oxidant sensitive dye 2′,7′-dichlorofluorescein diacetate in murine macrophage J774.1 cells. The antioxidants N-acetylcysteine and pyrrolidinedithiocarbamic acid abolished the hypoxic activation of NF-κB, TNF-α gene transcription, and increases in ROS levels. Rotenone, an inhibitor of mitochondrial complex I, abolished the increase in ROS signal, the activation of NF-κB, and TNF-α gene transcription during hypoxia. LPS stimulated NF-κB and TNF-α gene transcription but not ROS generation in J774.1 cells. Rotenone, pyrrolidinedithiocarbamic acid, and N-acetylcysteine had no effect on the LPS stimulation of NF-κB and TNF-α gene transcription, indicating that LPS activates NF-κB and TNF-α gene transcription through a ROS-independent mechanism. These results indicate that mitochondrial ROS are required for the hypoxic activation of NF-κB and TNF-α gene transcription, but not for the LPS activation of NF-κB.
Multiple organ system failure (MOSF)3 is the leading cause of mortality in medical and surgical intensive care units (1). A salient feature of MOSF is the inappropriate regulation of inflammatory cytokines and the failure of systemic host defense (2). A leading cause of MOSF is systemic sepsis, which is associated with cardiovascular dysfunction through its effects on the myocardium, endothelium, and vascular smooth muscle (3). Sepsis-induced MOSF can be initiated by circulating LPS (3) derived from the outer membrane of Gram-negative bacteria (4, 5). LPS activates the transcription and subsequent release of cytokines, including TNF-α, through a receptor- mediated signaling pathway (6, 7). TNF-α under physiological conditions has an important role in maintaining hemodynamics, host defense, and repair of tissue injury (8). However, during a systemic inflammatory response to LPS, the unregulated release of TNF-α into the circulation results in circulatory dysfunction, increased endothelial permeability, and inflammation in multiple organs, including the lung (9, 10, 11). Lung gas exchange failure and circulatory disturbances can lead to hypotension and tissue hypoxia, which can exacerbate the LPS-induced release of TNF-α and result in worsening of the sepsis syndrome (12, 13, 14).
The transcription factor NF-κB mediates the LPS-induced release of TNF-α at the cellular level (15). Hypoxia also stimulates NF-κB activation and TNF-α gene transcription (16, 17, 18, 19, 20). The activated form of NF-κB is a heterodimer, which usually consists of two proteins, a p65 subunit and a p50 subunit (21). Both subunits of NF-κB are members of the NF-κB/Rel family of transcription factors, which also includes c-Rel, RelB, and p52. The activity of NF-κB is regulated by an inhibitor, IκBα, which forms a complex with NF-κB in the cytoplasm and inhibits the nuclear localization of the dimer (22). When cells receive signals that activate NF-κB, IκBα is phosphorylated and degraded through an ubiquitin proteasome pathway (23). The degradation of IκBα triggers the translocation of NF-κB from the cytoplasm to the nucleus and activates the transcription of specific target genes. However, it is still unclear how diverse stimuli regulate IκBα phosphorylation and subsequent degradation resulting in NF-κB activation. One common mechanism for the activation of NF-κB proposes a requirement for an increase in reactive oxygen species (ROS). Hydrogen peroxide exposure can rapidly activate NF-κB, and the thiol reductant pyrrolidinedithiocarbamic acid (PDTC), an effective antioxidant, can block NF-κB activation in response to LPS, TNF-α, and active phorbol ester in a number of different cell lines (24, 25). The antioxidant N-actylcysteine (NAC) blocks activation of NF-κB by hypoxia (26). However, these previous reports did not measure endogenous ROS production or describe the source of ROS generation required for the activation of NF-κB by hypoxia or LPS (27). Recently, mitochondria have been described as a major source of ROS in response to specific stimuli that include hypoxia (28). In the present study, we tested whether ROS generated by the mitochondrial electron transport chain are required for the hypoxic and LPS activation of NF-κB and for the induction of TNF-α mRNA in the J774.1 murine macrophage cell line.
Materials and Methods
Cell culture and measurement of ROS
J774.1 cells were cultured on glass coverslips, 35-mm petri dishes, or T-75 flasks to 70–90% confluence in RPMI with penicillin (100 U/ml), streptomycin (100 μg/ml), and 5% heat-inactivated FCS (Life Technologies, Gaithersburg, MD). Hypoxic conditions were achieved by as previously described (28). ROS generation was assessed using the oxidant-sensitive probes 2′,7′-dichlorofluorescein diacetate (DCFH-DA, 10 μM, Molecular Probes, Eugene, OR) or dihydroethidium (DHE, 10 μM, Molecular Probes) (28).
Electrophoretic mobility shift assay (29)
Nuclear extracts were prepared from cells by lysing them in a hypotonic solution (10 mM KCl, 10 mM HEPES, protease cocktail inhibitors (Boehringer Mannheim, Indianapolis, IN), 1 mM PMSF, freshly added) and centrifuged at 14,000 rpm. Subsequently, the pellet was suspended in a solution containing 400 mM KCl, 20 mM HEPES, 25% (v/v) glycerol, 0.2 mM EDTA, and 1.5 mM MgCl2, pH 7.8. The solution was centrifuged to pellet the nuclei debris, and the proteins in the supernatant were used to perform Western blot analysis. To perform electrophoretic mobility shift assays, the supernatant was diluted with 2 volumes of a solution containing 20 mM HEPES, 25% glycerol (v/v), 0.2 mM EDTA, and 1.5 mM MgCl2, pH 7.8. The DNA binding reactions were performed at room temperature in buffer containing 20 mM HEPES, 100 mM KCl, 1 mM EDTA, 1 μg calf thymus DNA, 50,000 dpm labeled probe, 10% glycerol, and 5 μg nuclear extracts in a volume of 25 μl at pH 7.8. The oligonucleotide probe used in the binding reactions consisted of the sequence 5′-AGCTTCAGAGGGGACTTTCCGAGAGG-3′; 3′- AGTCTCCCCTGAAAGGCTCTCCAGCT-5′.
The probe was labeled with [α-32P]dGTP (800 Ci/mol) and [α-32P]dCTP (800 Ci/mol) by Klenow polymerase. The specificity of NF-κB DNA binding activity was determined by supershift analysis with p65 Abs. All reaction mixtures were electrophoresed on 4% nondenaturing polyacrylamide gel. Subsequently, the gels were dried and autoradiographed.
Northern analysis
Total cellular RNA (tcRNA) was isolated from J774.1 cells using the RNA isolation kit (Qiagen, Chatsworth, CA) according to the manufacturer’s protocol. Equal aliquots (10 μg) of each tcRNA sample were denatured and electrophoresed on a 1.2% agarose-formaldehyde gel. In preparation for transfer, the gel was soaked in 0.2 M NaOH for 15 min followed by 45 min in solution containing 1.5 M sodium chloride and 0.15 M sodium citrate. The gel was blotted by capillary transfer to Hybond-N nylon membrane (Amersham Pharmacia Biotech, Piscataway, NJ) for 24–36 h. After the transfer, RNA was cross-linked to the membrane under UV light. Prehybridization of the blots was conducted in prehybridization solution (Life Technologies), for 2–4 h at 65°C. Blots were then transferred to hybridization solution (1.0 M sodium chloride, 0.1 M sodium citrate, 0.01 M EDTA, pH 8.0; 5× Denhardt’s solution; 0.5% SDS; 100 μg/ml sheared, denatured salmon sperm DNA; Life Technologies), and denatured 32P-labeled TNF-α probe was added. Hybridization was allowed to proceed overnight (16–24 h) at 65°C. After hybridization, membranes were washed twice for 5 min at room temperature (0.3 M sodium chloride, 0.03 M sodium citrate, 0.1% SDS), twice for 20 min at 65°C (0.015 M sodium chloride, 0.0015 M sodium citrate, 0.1% SDS), once for 15 min at room temperature (0.015 M sodium chloride, 0.0015 M sodium citrate, 0.1% SDS), and once for 10 min at room temperature (0.3 M sodium chloride and 0.03 M sodium citrate). Northern blots were analyzed by autoradiography.
Preparation of 32P-labeled TNF-α probe
The probe for TNF-α was obtained by RT-PCR. The following primer sequences were extracted from the cDNA sequence for murine TNF-α (30): 5′GAGCAGCTGGAGTGGCTGCTGAG 3′(sense), and 5′ TAGACCTGCCCGGACTCCGC 3′(antisense). Using these primers and tcRNA isolated from LPS (100 ng/ml)-treated J774.1 cells as the template, a 379-bp fragment was amplified. For Northern hybridizations, the PCR product was gel purified, and 25–50 ng were 32P-labeled by random primer labeling using DNA Labeling Beads (dCTP) (Amersham Pharmacia Biotech).
Cytokine immunoassays
TNF-α was measured in supernatants of J774.1 cells by ELISA, as described by manufacturer’s directions (R&D Systems, Minneapolis, MN).
Results
ROS generation during LPS is dependent on the production of TNF-α
LPS has been shown to cause an increase in cellular oxidative stress (31). However, whether this increase in ROS reflects a direct response to LPS or an indirect response to LPS-induced cytokine release remains controversial (31). To assess whether LPS directly induces generation of ROS, murine macrophage J774.1 cells were placed in a flowthrough chamber on an inverted microscope under normoxic conditions (15% O2) in the presence of LPS (1 μg/ml) and DCFH-DA (10 μM) for 90 min. The DCFH-DA dye can be oxidized to the fluorescent compound 2′7′-dichlorofluorescein (DCF) by hydrogen peroxide within cells (32). LPS failed to increase the oxidation of the DCFH dye during 90 min, indicating no increase in ROS (Fig. 1,A). To assess whether prolonged exposure to LPS induces ROS, J774.1 cells were placed in 35-mm petri dishes and incubated with LPS at different concentrations in the presence of DCFH-DA. At various time points, the medium was aspirated, and the cells were lysed and centrifuged to remove debris. Afterward, the fluorescence in the supernatant was measured. LPS increased oxidation of DCFH during 6–18 h in a dose-dependent manner (Fig. 1,B). The increase in DCF fluorescence was abolished in the presence of the antioxidants NAC (500 μM) and PDTC (10 μM) (Fig. 2,A). Both antioxidants maintain reduced levels of glutathione, thereby enhancing the scavenging of hydrogen peroxide. To determine whether the LPS induction of ROS by LPS required de novo protein synthesis, J774.1 cells were incubated for 18 h in the presence of cycloheximide, a protein synthesis inhibitor. The LPS-induced increase in DCF fluorescence was attenuated in the presence of cycloheximide (25 μg/ml), indicating that LPS generation of ROS requires de novo protein synthesis (Fig. 2,A). Previous studies have shown that TNF-α increases generation of ROS (33). Indeed, TNF-α production progressively increased during 18 h (Fig. 2 B), and the LPS increase in DCF fluorescence was attenuated in the presence of TNF-α-neutralizing Ab. These data indicate that LPS does not increase ROS acutely (within 90 min), but LPS does induce ROS generation during 6–18 h by stimulating the secretion of TNF-α that acts in an autocrine fashion on the cell surface receptors.

A, DCF fluorescence in J774.1 cells exposed to LPS (1 μg/ml). J774.1 cells were placed in a flowthrough chamber, and at t = 10 min LPS was added to the perfusate. B, DCF fluorescence in J774.1 cells as a function of time and different concentrations of LPS.
A, DCF fluorescence in J774.1 cells exposed to LPS (1 μg/ml). J774.1 cells were placed in a flowthrough chamber, and at t = 10 min LPS was added to the perfusate. B, DCF fluorescence in J774.1 cells as a function of time and different concentrations of LPS.

A, DCF fluorescence in J774.1 cells exposed to LPS (1 μg/ml) for 18 h in the presence of NAC (500 μM), PDTC (10 μM), cycloheximide (CHX, 25 μg/ml), and a TNF-α-neutralizing Ab (TNF-α Ab). B, TNF-α release from J774.1 macrophages exposed to LPS (1 μg/ml) for 6, 12, or 18 h (n = 4). C, DCF fluorescence in J774.1 cells exposed to LPS (1 μg/ml) in the presence of apocynin (300 μM), DPI (10 μM), rotenone (1.0 μg/ml), and antimycin A (1.0 μg/ml) for 18 h.
A, DCF fluorescence in J774.1 cells exposed to LPS (1 μg/ml) for 18 h in the presence of NAC (500 μM), PDTC (10 μM), cycloheximide (CHX, 25 μg/ml), and a TNF-α-neutralizing Ab (TNF-α Ab). B, TNF-α release from J774.1 macrophages exposed to LPS (1 μg/ml) for 6, 12, or 18 h (n = 4). C, DCF fluorescence in J774.1 cells exposed to LPS (1 μg/ml) in the presence of apocynin (300 μM), DPI (10 μM), rotenone (1.0 μg/ml), and antimycin A (1.0 μg/ml) for 18 h.
Intracellular generation of ROS can originate from a number of potential sources, including NADPH oxidase and complex III within the mitochondrial electron transport chain (34). Diphenyleneidonium (DPI) is an inhibitor of a wide range of flavoproteins, including NADPH oxidase and complex I within the mitochondrial electron transport chain (35). DPI (10 μM) attenuated the increase in DCF fluorescence in response to LPS (Fig. 2,C). Apocynin (300 μM), a NADPH oxidase inhibitor, did not abolish the increase in DCF fluorescence in response to prolonged exposure to LPS (Fig. 2,C). Rotenone (1 μg/ml), an inhibitor of electron transport that acts at complex I, abolished the increase in DCF fluorescence in response to LPS (Fig. 2 C). By contrast, antimycin A (1 μg/ml), an inhibitor of electron transport at the downstream end of complex III, did not alter the increase in DCF fluorescence. These results indicate that the site of mitochondrial ROS generation during prolonged exposure is between complex I and complex III.
Hypoxia increases mitochondrial ROS generation
Intracellular ROS generation during hypoxia was measured using DCFH-DA in J774.1 cells placed in a flowthrough chamber on an inverted microscope. J774.1 cells demonstrated an increase in DCF fluorescence in response to hypoxia (1.5% O2) (Fig. 3,A). The antioxidants NAC or PDTC and DPI abolished the increase in DCF signal in response to hypoxia (Fig. 3,B). Previous studies in Hep3B cells showed that hypoxia increases superoxide generation as a result of electron flux through mitochondrial complex III (28). In J774.1 cells, rotenone abolished the increase in DCF signal observed during hypoxia. (Fig. 3,B). By contrast, antimycin A further augmented the hypoxia-induced increase in DCF fluorescence (Fig. 3,B). The NADPH oxidase inhibitor apocynin did not abolish the hypoxic increase in DCF signal (Fig. 3,B). To further demonstrate that hypoxia stimulates mitochondrial ROS production, J774.1 cells were incubated with DHE in the presence of hypoxia and various inhibitors. DHE undergoes oxidation to fluorescent ethidium in the presence of superoxide (36). Fig. 3 C shows that hypoxia increases ethidium fluorescence, indicating an increase in superoxide generation. The hydrogen peroxide-scavenging antioxidants NAC and PDTC did not affect the hypoxic increase in ethidium fluorescence. By contrast, rotenone abolished the increase in superoxide generation. Collectively, these results demonstrate that hypoxia directly induces mitochondrial ROS in J774.1 cells.

A, DCF fluorescence in J774.1 cells exposed to hypoxia (1.5% O2). B, DCF fluorescence in J774.1 cells exposed to hypoxia (1.5% O2) for 2 h in the presence of NAC (500 μM), PDTC (10 μM), rotenone (ROT, 1.0 μg/ml), DPI (10 μM), antimycin A (ANTI, 1.0 μg/ml), and apocynin (APO, 300 μM). C, Oxidation of DHE in J774.1 cells exposed to hypoxia (1.5% O2) for 2 h in the presence of NAC (500 μM), PDTC (10 μM), rotenone (1.0 μg/ml), DPI (10 μM), antimycin A (1.0 μg/ml), and apocynin (300 μM).
A, DCF fluorescence in J774.1 cells exposed to hypoxia (1.5% O2). B, DCF fluorescence in J774.1 cells exposed to hypoxia (1.5% O2) for 2 h in the presence of NAC (500 μM), PDTC (10 μM), rotenone (ROT, 1.0 μg/ml), DPI (10 μM), antimycin A (ANTI, 1.0 μg/ml), and apocynin (APO, 300 μM). C, Oxidation of DHE in J774.1 cells exposed to hypoxia (1.5% O2) for 2 h in the presence of NAC (500 μM), PDTC (10 μM), rotenone (1.0 μg/ml), DPI (10 μM), antimycin A (1.0 μg/ml), and apocynin (300 μM).
LPS and hypoxia increase NF-κB DNA binding activity independent of de novo protein synthesis
LPS and hypoxia independently have been shown to activate NF-κB DNA binding (17). To demonstrate this in J774.1 cells, NF-κB DNA-binding activity was assessed using an electrophoretic mobility shift assay. Nuclear extracts prepared from J774.1 cells exposed to either LPS (100 ng/ml) or hypoxia (1.5% O2) for 2 h contained a protein complex capable of binding a DNA probe containing a NF-κB binding site (Fig. 4). LPS and hypoxia both demonstrated bands containing the p65 subunit in the protein complex as demonstrated by supershift analysis (Fig. 4). Neither the hypoxia nor the LPS-induced NF-κB DNA-binding activity was abolished in the presence of cycloheximide (25 μg/ml) (Fig. 4). The hypoxia-induced NF-κB DNA binding activity was less evident than the LPS-induced NF-κB DNA-binding activity. Hypoxia did not augment the LPS-induced NF-κB DNA-binding activity. These results indicate that both hypoxia and LPS independently stimulate NF-κB DNA binding activity without de novo protein synthesis.

NF-κB DNA-binding activity in nuclear extracts from J774.1 cells exposed to hypoxia (1.5% O2) or to LPS (100 ng/ml) in the presence of cycloheximide (CHX, 25 μg/ml) for 2 h. An Ab against p65 subunit of NF-κB was added to demonstrate specificity.
NF-κB DNA-binding activity in nuclear extracts from J774.1 cells exposed to hypoxia (1.5% O2) or to LPS (100 ng/ml) in the presence of cycloheximide (CHX, 25 μg/ml) for 2 h. An Ab against p65 subunit of NF-κB was added to demonstrate specificity.
ROS-independent induction of NF-κB DNA binding and TNF-α mRNA during LPS
LPS activation of NF-κB DNA-binding activity has been suggested to require an increase in ROS (37, 38). These reports relied on the observation that antioxidants such as PDTC or NAC abolished the LPS activation of NF-κB DNA-binding activity. However, our current observations indicated that LPS does not directly increase ROS generation. To clarify the role of ROS in LPS induction of NF-κB DNA-binding activity, J774.1 cells were exposed to LPS (100 ng/ml) for 2 h at various concentrations of PDTC. Fig. 5a shows that 500 μM PDTC was required to abolish NF-κB DNA-binding activity. Yet PDTC is an effective antioxidant at concentrations of 10 μM. To clarify the role of ROS in LPS-induced transcription, NF-κB DNA-binding activity and TNF-α mRNA levels were examined in J774.1 cells. The antioxidants NAC (500 μM) and PDTC (10 μM) failed to abolish NF-κB DNA binding activity and TNF-α mRNA levels in response to LPS (Fig. 5, B and C). The mitochondrial inhibitors rotenone and antimycin A did not affect NF-κB DNA-binding activity or TNF-α mRNA levels in response to LPS (Fig. 5, B and C). Apocynin, a NADPH oxidase inhibitor, and DPI, an inhibitor of a wide range of flavoproteins, also did not alter LPS-induced NF-κB DNA-binding activity and TNF-α mRNA levels (Fig. 5, B and C). These results indicate that LPS activates NF-κB DNA-binding activity independently of an increase in ROS.

A, NF-κB DNA-binding activity in nuclear extracts from J774.1 cells exposed to 2 h of LPS (100 ng/ml) during normoxia in the presence of different concentrations of PDTC for 2 h. B, NF-κB DNA-binding activity in nuclear extracts from J774.1 cells exposed to LPS (100 ng/ml) in the presence of NAC (500 μM), PDTC (10 μM), apocynin (300 μM), antimycin A (1 μg/ml), rotenone (1 μg/ml), and DPI (10 μM) for 2 h. C, Northern blot analysis of TNF-α mRNA levels from J774.1 cells exposed to LPS (100 ng/ml) in the presence of NAC (500 μM), PDTC (10 μM), apocynin (300 μM), antimycin A (1 μg/ml); rotenone (1 μg/ml), and DPI (10 μM) for 6 h.
A, NF-κB DNA-binding activity in nuclear extracts from J774.1 cells exposed to 2 h of LPS (100 ng/ml) during normoxia in the presence of different concentrations of PDTC for 2 h. B, NF-κB DNA-binding activity in nuclear extracts from J774.1 cells exposed to LPS (100 ng/ml) in the presence of NAC (500 μM), PDTC (10 μM), apocynin (300 μM), antimycin A (1 μg/ml), rotenone (1 μg/ml), and DPI (10 μM) for 2 h. C, Northern blot analysis of TNF-α mRNA levels from J774.1 cells exposed to LPS (100 ng/ml) in the presence of NAC (500 μM), PDTC (10 μM), apocynin (300 μM), antimycin A (1 μg/ml); rotenone (1 μg/ml), and DPI (10 μM) for 6 h.
Hypoxia induction of NF-κB DNA binding and TNF-α mRNA requires an increase in ROS
Previous studies have shown that NAC abolishes hypoxia-induced NF-κB DNA-binding activity (26). In the present study, the antioxidants NAC (500 μM) and PDTC (10 μM) abolished NF-κB DNA-binding activity and TNF-α mRNA levels in J774.1 cells exposed to hypoxia (Fig. 6). To determine the dependence of the hypoxic response on mitochondrial ROS production, J774.1 cells were exposed to hypoxia in the presence of rotenone or antimycin A. Rotenone abolished NF-κB DNA-binding activity in response to hypoxia, whereas antimycin A maintained the hypoxic induction of NF-κB DNA binding activity (Fig. 6). These results are consistent with rotenone decreasing the hypoxic increase in ROS generation by limiting electron flux into complex III, the site of mitochondrial ROS generation. Antimycin A also inhibits electron transport, but at a site downstream of the location of ROS generation, thereby augmenting ROS generation. DPI abolished NF-κB DNA-binding activity and TNF-α mRNA levels in response to hypoxia. By contrast, apocynin had no effect on the hypoxic induction of NF-κB DNA-binding activity and TNF-α mRNA levels. These results indicate that the hypoxic induction of NF-κB DNA-binding activity and TNF-α mRNA levels requires mitochondrial ROS generation.
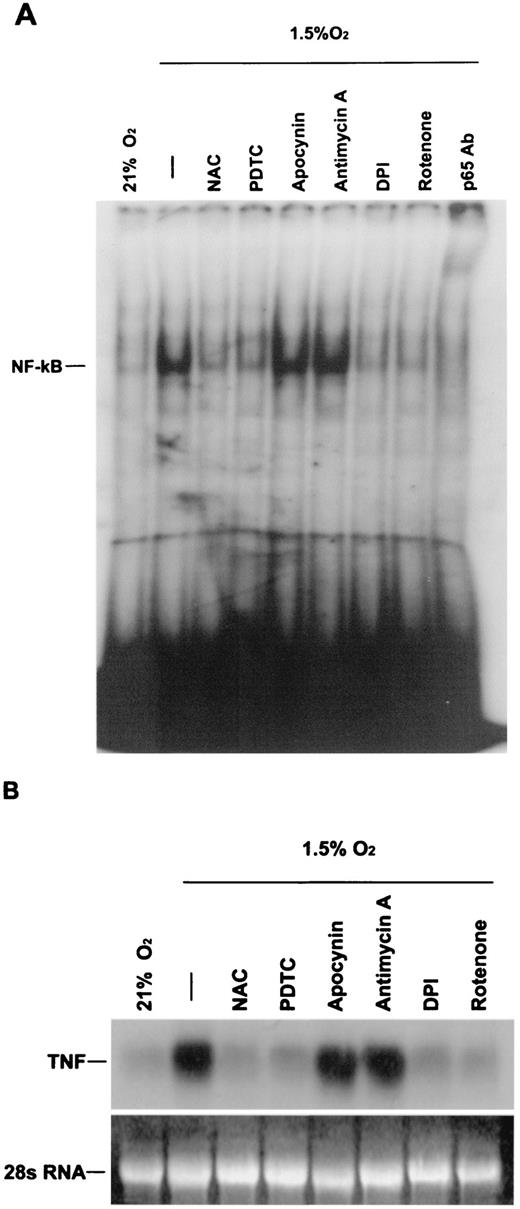
A, NF-κB DNA-binding activity in nuclear extracts from J774.1 cells exposed to hypoxia (1.5% O2) in the presence of the antioxidants NAC (500 μM) and PDTC (10 μM), apocynin (300 μM), antimycin (1 μg/ml), DPI (10 μM) and rotenone (1 μg/ml), for 2 h. B, Northern blot analysis of TNF-α mRNA levels from J774.1 cells exposed to hypoxia (1.5% O2) in the presence of the antioxidants NAC (500 μM) and PDTC (10 μM), apocynin (300 μM), antimycin (1 μg/ml), DPI (10 μM), and rotenone (1 μg/ml) for 6 h.
A, NF-κB DNA-binding activity in nuclear extracts from J774.1 cells exposed to hypoxia (1.5% O2) in the presence of the antioxidants NAC (500 μM) and PDTC (10 μM), apocynin (300 μM), antimycin (1 μg/ml), DPI (10 μM) and rotenone (1 μg/ml), for 2 h. B, Northern blot analysis of TNF-α mRNA levels from J774.1 cells exposed to hypoxia (1.5% O2) in the presence of the antioxidants NAC (500 μM) and PDTC (10 μM), apocynin (300 μM), antimycin (1 μg/ml), DPI (10 μM), and rotenone (1 μg/ml) for 6 h.
Hypoxia augments LPS induction of TNF-α
TNF-α mRNA levels and TNF-α release were measured in J774.1 cells exposed to hypoxia, LPS, and LPS plus hypoxia. Hypoxia stimulated TNF-α mRNA levels and TNF-α release from J774.1 cells but ∼10-fold less than LPS (Fig. 7,A). Interestingly, hypoxia augmented LPS induction of TNF-α mRNA levels and TNF-α release (Fig. 7,B). NAC and PDTC abolished both the response to hypoxia alone and the augmentation of LPS-induced TNF-α mRNA levels and TNF-α release from J774.1 cells by hypoxia (Fig. 7, A and B). Rotenone and DPI both abolished the hypoxic augmentation of LPS-induced TNF-α mRNA levels. Antimycin A and apocynin did not alter the hypoxic augmentation of LPS-induced TNF-α mRNA levels. These results are consistent with the hypothesis that the hypoxic signaling pathway requires an increase in mitochondrial ROS.

A, TNF-α release from J774.1 macrophages exposed to hypoxia (1.5% O2) and LPS (1 μg/ml) for 18 h in the presence of NAC (500 μM) and PDTC (10 μM); n = 4. B, Northern blot analysis of RNA isolated from J774.1 cells exposed to normoxia (21% O2), hypoxia (1.5% O2), LPS (100 ng/ml), and LPS + hypoxia for 6 h. C, Northern blot analysis of RNA isolated from J774.1 cells exposed to LPS (100 ng/ml) + hypoxia (1.5% O2) in the presence of NAC (500 μM), PDTC (10 μM), apocynin (300 μM), antimycin A (1 μg/ml), DPI (10 μM), and rotenone (1 μg/ml) for 6 h. Note: exposures for LPS Northern blots were for a shorter duration than for the hypoxia Northern blots (Fig. 6 B).
A, TNF-α release from J774.1 macrophages exposed to hypoxia (1.5% O2) and LPS (1 μg/ml) for 18 h in the presence of NAC (500 μM) and PDTC (10 μM); n = 4. B, Northern blot analysis of RNA isolated from J774.1 cells exposed to normoxia (21% O2), hypoxia (1.5% O2), LPS (100 ng/ml), and LPS + hypoxia for 6 h. C, Northern blot analysis of RNA isolated from J774.1 cells exposed to LPS (100 ng/ml) + hypoxia (1.5% O2) in the presence of NAC (500 μM), PDTC (10 μM), apocynin (300 μM), antimycin A (1 μg/ml), DPI (10 μM), and rotenone (1 μg/ml) for 6 h. Note: exposures for LPS Northern blots were for a shorter duration than for the hypoxia Northern blots (Fig. 6 B).
Discussion
TNF-α acts as a signaling element in the pathophysiology of the systemic inflammatory response in critically ill patients. LPS is thought to be the principal causative agent for the regulated release of cytokines associated with sepsis and is derived from the outer membrane of Gram-negative bacteria. LPS released from bacteria in blood or tissues interacts with serum factors, such as LPS-binding protein, which interacts with the membrane receptor CD14 on cells. Binding to CD14 leads to the activation of kinases that result in NF-κB activation (39, 40). How does CD14 binding lead to kinase activation? One suggestion has been that a burst of intracellular ROS is generated, leading to the activation of kinases and thus NF-κB (41, 42). The evidence for cellular oxidative signaling involvement in the activation of NF-κB is based on three observations: 1) antioxidants such as PDTC and NAC abolish LPS-induced activation of NF-κB (37, 38, 34); 2) in vitro administration of H2O2 to cells stimulates activation of NF-κB (43); 3) NAC blocks activation of NF-κB in animal models of ARDS and has been reported to improve lung function in patients with adult respiratory distress syndrome (44, 45, 46). One concern regarding these studies is that unusually high concentrations of NAC (>10 mM) and PDTC (>100 μM) were required to abolish NF-κB activation. Moreover, there was no direct evidence that LPS leads to an increase in ROS generation over the time period required to observe NF-κB activation. In the present study, we examined the relationship between cellular oxidative stress and NF-κB activation in response to LPS. The data indicate that LPS does not require an increase in ROS generation for NF-κB activation and the subsequent expression of TNF-α mRNA. There was no observable increase in ROS generation over the time period required to observe NF-κB activation. The antioxidant PDTC abolished LPS-induced NF-κB activity at 500 μM. However, PDTC is an effective ROS scavenger at the minimal concentration of 10 μM. Thus, at higher doses it is likely that PDTC is interfering in NF-κB DNA-binding activity in response to LPS rather than through the attenuation of ROS generation. PDTC may target kinase activation. Many kinases contain cysteine residues that are likely to be redox sensitive to a thiol reductant such as PDTC. The antioxidant NAC also did not affect LPS activation of NF-κB or subsequent TNF-α expression at an antioxidant dose (500 μM) that was effective in abolishing the oxidant signaling during hypoxia as indicated by the DCFH dye. Moreover, LPS-induced NF-κB activity and TNF-α mRNA expression were not altered in the presence of specific inhibitors of NADPH oxidase and mitochondrial electron transport, which suggests that key ROS-generating systems did not participate in the signaling pathway.
Previous studies have indicated that macrophages challenged with LPS exhibit an increase in oxidative stress (47). This effect could have been mediated by release of TNF-α or nitric oxide. Because LPS did not show an increase in oxidative signaling during the acute period (2 h) required to elicit NF-κB activation, we tested whether prolonged exposure of LPS would increase ROS generation. The ROS measurements in response to LPS during 2 h were done in a flowthrough chamber; therefore, any released cytokines would have been carried away rather than allowed to accumulate. Longer exposure to LPS (12–18 h) in a static petri dish did elicit an increase in ROS generation. This response was due to the autocrine effect of TNF-α on the cell membrane receptor because the ROS increase was abolished in the presence of TNF-α-neutralizing Ab. Furthermore, mitochondrial complex I inhibitors abolished the LPS induced increase in ROS during 18 h. Previous reports have indicated that TNF-α increases ROS at complex III of the mitochondrial electron transport chain (48). Thus, LPS does not directly induce ROS, but over time LPS induces ROS through de novo synthesis of TNF-α acting on complex III within mitochondrial electron transport chain (Fig. 8).

Effects of LPS and hypoxic activation of NF-κB and TNF-α mRNA. LPS activates NF-κB and TNF-α mRNA expression. Subsequently, TNF-α increases mitochondrial ROS through an autocrine effect of TNF-α on a cell membrane receptor. Hypoxia directly induces mitochondrial ROS, which subsequently activate NF-κB and TNF-α mRNA expression.
Effects of LPS and hypoxic activation of NF-κB and TNF-α mRNA. LPS activates NF-κB and TNF-α mRNA expression. Subsequently, TNF-α increases mitochondrial ROS through an autocrine effect of TNF-α on a cell membrane receptor. Hypoxia directly induces mitochondrial ROS, which subsequently activate NF-κB and TNF-α mRNA expression.
Lung gas exchange failure and cardiovascular dysfunction during sepsis can lead to tissue hypoxia. Hypoxia alone has been shown to activate NF-κB and release of cytokines including TNF-α. In the present study, we demonstrate that hypoxia increases NF-κB DNA-binding activity and TNF-α mRNA levels. The hypoxic activation of NF-κB and subsequent TNF-α mRNA gene transcription require increases in mitochondrial ROS generation. Previous studies have reported that mitochondrial complex III can function as an important site of superoxide generation during hypoxia (28). Evidence for mitochondrial ROS regulation of NF-κB during hypoxia rests on the following observations. Hypoxia increased NF-κB DNA-binding activity and ROS generation in the presence of cycloheximide, indicating that hypoxia does not require de novo factors such as TNF-α. The antioxidants NAC and PDTC abolished the hypoxic increase in DCF fluorescence, NF-κB DNA-binding activity, and TNF-α mRNA levels. The specific mitochondrial complex I inhibitor rotenone abolished the increase in DCF fluorescence, NF-κB DNA-binding activity and TNF-α mRNA levels during hypoxia. By contrast, the mitochondrial complex III inhibitor antimycin A did not abolish the increase in ROS signaling, NF-κB DNA-binding activity, and TNF-α mRNA levels during hypoxia. The observation that rotenone abolished ROS signaling, NF-κB activation, and TNF-α mRNA expression during hypoxia whereas antimycin A preserved these responses during hypoxia indicates that mitochondrial ATP is not required for the response, because both inhibitors block electron transport and abolish oxidative phosphorylation. These results indicate that mitochondrial ROS are required for the activation of NF-κB DNA-binding activity and TNF-α mRNA levels during hypoxia (Fig. 8).
Under physiological conditions, low levels of cytokines have important roles in maintaining vascular function, in host defense, and in controlling the repair of tissue injury. However, during systemic sepsis or in response to experimental LPS challenge, the uncontrolled release of cytokines into the circulation contributes to the circulatory dysfunction and may precipitate the development of organ injury in multiple organs. Why is the activation of NF-κB by hypoxia potentially important in this scheme? The activation of NF-κB by hypoxia and the subsequent local cytokine release is part of the normal repair of local tissue injury and thus an adaptive function. For example, localized tissue hypoxia may contribute to the activation of NF-κB and local cytokine release, which could be important in controlling the infectious process and enhancing wound healing. By contrast, during systemic inflammatory states associated with sepsis, hypoxia could augment the excessive release of cytokines that are already stimulated by LPS. Our data support this hypothesis by demonstrating that LPS stimulates TNF-α release 10-fold more than hypoxia alone in macrophages. However, hypoxia significantly augments the LPS-induced release of TNF-α. Antioxidants attenuated the hypoxic augmentation of LPS-induced release of TNF-α, as did inhibitors of the proximal regions of the mitochondrial electron transport chain. Interestingly, hypoxia did not augment LPS-induced NF-κB DNA-binding activity, although hypoxia did augment TNF-α mRNA expression. This suggests that hypoxia may activate transcription factors other than NF-κB that play a role in the hypoxic amplification of LPS induction of TNF-α mRNA levels. Collectively, these studies reveal that hypoxic amplification of LPS gene transcription relies on signaling pathways that involve mitochondrial ROS.
Footnotes
This work was supported by Grants HL32646 and HL 35440. N.S.C was supported by an American Lung Association Research Fellowship grant.
Abbreviations used in this paper: MOSF, multiple organ system failure; DCF, 2′7′-dichlorofluorescein; DCFH-DA, 2′,7′-dichlorofluorescin diacetate; DHE, dihydroethidium; ROS, reactive oxygen species; NAC, N-acetylcysteine; PDTC, pyrrolidinedithiocarbamic acid; tcRNA, total cellular RNA; DPI, diphenyleneidonium.
