

Abstract
Wiskott-Aldrich syndrome protein (WASP)-deficient T cells exhibit defects in IL-2 production that are widely believed to stem from primary defects in actin remodeling and immune synapse formation. Surprisingly, however, we find that WASP-deficient T cells responding to Ag-specific APCs polymerize actin and organize talin and PKCθ normally, forming an immune synapse that is stable for at least 3 h. At low doses of peptide, WASP-deficient T cells show less efficient talin and PKCθ polarization. Thus, although WASP may facilitate immune synapse formation at low peptide concentrations, WASP is not required for this process. Defects in IL-2 production are observed even under conditions in which immune synapse formation proceeds normally, suggesting that the role of WASP in regulating IL-2 production is independent of its role in immune synapse formation.
Early T cell signaling events trigger the organization of specific molecules at the (IS),3 a process that is closely correlated with productive T cell activation (1, 2). TCR and PKCθ localize to the center of the IS, or central supramolecular activation cluster (C-SMAC), whereas LFA-1 and talin segregate to the peripheral region (P-SMAC) (3). F-actin polymerizes at the IS and the microtubule organizing center (MTOC) reorients toward this site. Perturbation of actin dynamics inhibits TCR signaling, conjugate formation, organization of the IS, and IL-2 production (1). It is widely believed that IS formation is driven at least in part by actin-dependent processes including actin polymerization, lipid raft dynamics, and myosin-dependent molecular movements. However, the underlying molecular mechanisms are poorly understood.
One protein frequently proposed to regulate IS architecture is the Wiskott-Aldrich syndrome protein (WASP) (1, 4, 5, 6). Mutations in WASP cause Wiskott-Aldrich syndrome (WAS), an X-linked recessive disorder characterized by immunodeficiency, eczema, and thrombocytopenia (4, 7). T cells from WAS patients and WASP-deficient mice exhibit defects in actin polymerization, TCR capping, proliferation, and IL-2 production in response to anti-CD3 stimulation (4, 5, 7, 8). Dominant mutants of the WASP-activating protein Cdc42 have been reported to interfere with both MTOC and actin polarization in T cells (9), and T cells from WAS patients lack the ability to localize lipid rafts to the IS (10). These findings strongly suggest that WASP plays an important role in T cell activation through regulation of cytoskeletal dynamics.
WASP and its ubiquitous homolog N-WASP are multidomain proteins with numerous binding partners (11). The C-terminal verpolin/cofilin/acidic domain of WASP proteins binds the actin nucleation complex Arp2/3, activating formation of branched actin structures at the cell cortex. WASP proteins exist in an autoinhibited conformation, which is relieved on interaction with the Rho family GTPase Cdc42 and PI (4,5)-bisphosphate. We have previously shown that TCR engagement induces the accumulation of Cdc42-GTP and WASP at the IS (12), leading to the conformational change associated with WASP activation (13, 14). There is also evidence that WASP can be activated at the IS via tyrosine phosphorylation (15), creating binding sites for Src family kinases and other SH2 domain-containing proteins (16). WASP is recruited to the IS through interactions of its proline-rich domain with Nck or PSTPIP1 (12, 13, 17), and interacts through its N-terminal WASP homology 1 (WH1) domain with WASP-interacting protein (WIP; Ref.18). Many WAS mutations lie in the WH1 domain, indicating that WIP binding is important for WASP function (7, 19). WASP-WIP interaction is regulated upon T cell activation (20), and WIP-deficient T cells exhibit profound defects in actin remodeling and cytokine production (21).
In at least some experimental systems, WASP-deficient T cells form conjugates with APCs normally (22, 23), suggesting that not all actin-dependent aspects of T cell activation are mediated by WASP. We tested the ability of WASP-deficient T cells to polarize cytoskeletal proteins and IS markers under conditions in which conjugate production is unperturbed. Surprisingly, we found that these cells could efficiently polarize MTOC, actin, PKCθ, and talin to the IS. When stimulated with low doses of peptide, WASP-deficient T cells showed diminished polarization of PKCθ and talin. However, defects in IL-2 production were observed at all peptide doses, indicating that the requirement for WASP in signaling IL-2 production may be independent of its role in the formation of the IS.
Materials and Methods
Cells and reagents
AND TCR transgenic (Tg) and WASP knockout mice were from The Jackson Laboratory (Bar Harbor, ME). C3H mice were from the National Cancer Institute (Frederick, MD). Cells were cultured in DMEM-10% FCS as described previously (24). To generate T cell blasts, lymph node T cells from AND TCR Tg WASP+/− and WASP−/Y mice were cocultured with irradiated C3H splenocytes, 6.67 μg/ml DASP peptide (moth cytochrome c (MCC)86–90; MCC94–103), and 1 μg/ml anti-CD28. From 7 to 14 days after activation, live cells were recovered by Ficoll-Hypaque (Sigma-Aldrich, St. Louis, MO) gradient separation. Anti-cytoplasmic NFAT Abs were from Affinity Bioreagents (Golden, CO). Anti-lamin A, rabbit-anti-PKCθ (C-18), and goat anti-talin (C-20) were from Santa Cruz Biotechnology (Santa Cruz, CA); anti-FLAG Ab was from Kodak (Rochester, NY); anti-tubulin Ab DM1A was from Neomarkers (Fremont, CA). Fluorescent secondary Abs were from Jackson ImmunoResearch Laboratories (West Grove, PA). Rhodamine-phalloidin was from Molecular Probes (Eugene, OR).
Conjugation and immunofluorescence microscopy
Cell conjugations, immunofluorescence microscopy, and quantitation of actin and MTOC polarization were done as described previously (25), and polarization of PKCθ and talin was analyzed in the same way. To minimize bias in quantitive analyses, conjugates were identified in the differential interference contrast image as small T cells contacting larger B cells, regardless of presence or absence of Ag, cell morphology, or protein distribution. Deconvolution was performed using Openlab version 3.1.1 (Improvision, Coventry, U.K.) or SlideBook 3.0 (I3, Denver, CO). Z-series images were processed by nearest neighbor deconvolution and rendered in three dimensions using Slidebook. The length of the IS and T cell circumference were measured using NIH Image.
NFAT translocation assays
Lymph node cells were purified via nylon wool and were unactivated, incubated with beads (Dynal Biotech, Great Neck, NY) coated with 1 μg/ml 2C11 and 1 μg/ml CD28, uncoated beads, or 10 ng/ml PMA and 0.4 ng/ml ionomycin. Cells were then lysed in 10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM EDTA (pH 8), 0.1 mM EGTA (pH 8), 0.4% Nonidet P-40 on ice for 15 min. The nuclear pellet was extracted with 20 mM HEPES (pH 7.9), 0.4M NaCl, 1 mM EDTA, and 1 mM EGTA, vortexed for 2 min at 4°C, and clarified by centrifugation to generate the nuclear fraction. Samples were separated by SDS-PAGE, and proteins were detected using standard Western blotting protocols. Images of the chemiluminescent signal were captured using the Fujifilm LAS 3000 system (Stamford, CT) and quantitated using Fujifilm Image Gauge version 4.0. Relative NFATc levels were calculated by [NFATc signal]/[lamin A signal] to normalize for loading. The fold increase in nuclear NFATc was calculated by [corrected NFATc signal]/[corrected NFATc signal in the unactivated control].
IL-2 ELISA
T cells (2 × 105) from WASP+/− and WASP−/Y mice were cultured with 2×105 irradiated splenocytes from C3H spleens (2500 rad) and DASP peptide. Culture supernatants were analyzed using an IL-2 ELISA kit (BD Pharmingen, San Diego, CA) according to the manufacturer’s directions.
Results
WASP is required for IL-2 production in response to antigenic stimulation
WASP-deficient T cells show defects in IL-2 production in response to anti-CD3 Ab (4). To assess IL-2 responses to Ag-specific APCs, WASP-deficient mice were bred onto the AND TCR Tg background. This TCR recognizes I-Ek molecules presenting MCC88–103 peptide or its superagonist variant DASP (26, 27). When stimulated with splenocytes, naive WASP-deficient T cells showed defects in IL-2 production at all peptide doses (Fig. 1,A). Previously activated T cells showed similar defects using either splenocytes (Fig. 1,B) or the B cell line CH12 (Fig. 1,C) as APCs. In addition to showing defects in IL-2 production, WASP-deficient T cells showed diminished translocation of NFATc into the nuclear fraction (Fig. 1 D). These results are consistent with previous studies showing that WASP-deficient cells exhibit normal MAP kinase signaling, but partial defects in Ca2+ signaling and activation of an NFAT-reporter construct (5, 15). We conclude that the defect in NFAT translocation is the likely basis for the defective IL-2 production.
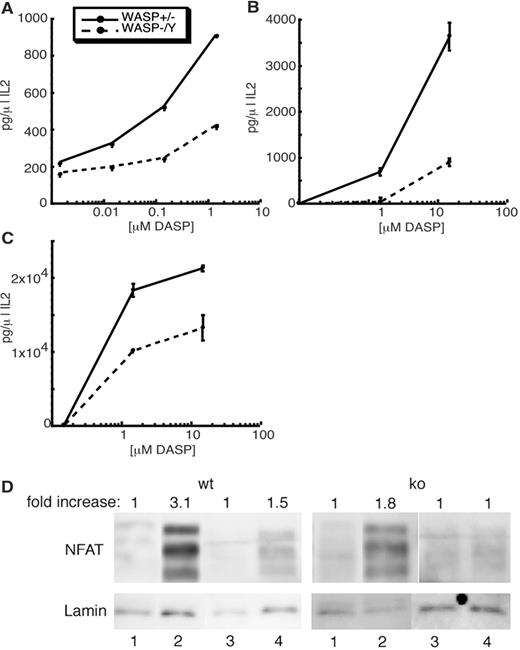
WASP-deficient T cells have a defect in IL-2 production and NFATc translocation in response to T cell stimulation. Naive (A and D) or previously activated (B and C) WASP+/− or WASP−/Y lymph node T cells were incubated for 24 h with C3H splenocytes (A and B) or CH12 B cells (C) presenting the indicated concentrations of DASP peptide. Supernatants were analyzed by ELISA for IL-2 production. D, NFATc nuclear translocation in cells that were unactivated (lanes 1), activated with PMA/ionomycin (lanes 2), mock-activated with uncoated beads (lanes 3), or activated with anti-CD3/anti-CD28-coated beads (lanes 4). In each case, nuclear fractions were separated and analyzed by SDS-PAGE, and NFATc and Lamin A were detected by Western blot analysis. Fold increase in nuclear NFATc over the unactivated control was determined using lamin A as a loading control, as described in Materials and Methods.
WASP-deficient T cells have a defect in IL-2 production and NFATc translocation in response to T cell stimulation. Naive (A and D) or previously activated (B and C) WASP+/− or WASP−/Y lymph node T cells were incubated for 24 h with C3H splenocytes (A and B) or CH12 B cells (C) presenting the indicated concentrations of DASP peptide. Supernatants were analyzed by ELISA for IL-2 production. D, NFATc nuclear translocation in cells that were unactivated (lanes 1), activated with PMA/ionomycin (lanes 2), mock-activated with uncoated beads (lanes 3), or activated with anti-CD3/anti-CD28-coated beads (lanes 4). In each case, nuclear fractions were separated and analyzed by SDS-PAGE, and NFATc and Lamin A were detected by Western blot analysis. Fold increase in nuclear NFATc over the unactivated control was determined using lamin A as a loading control, as described in Materials and Methods.
WASP is not required for actin or MTOC polarization
Because WASP-deficient T cells can efficiently form conjugates with APCs (22, 23), we asked whether actin remodeling occurs normally in these conjugates. T cells from wild-type (wt) or WASP-deficient mice were conjugated to peptide-pulsed B cells, and the accumulation of F-actin at the IS was evaluated. Surprisingly, WASP-deficient T cells accumulated F-actin normally (Fig. 2,A). No difference was observed in the intensity of F-actin labeling or the length of the contact site, calculated as a percentage of T cell circumference (30% for wt vs 28% for WASP−/Y T cells). Moreover, the frequency of conjugates showing F-actin at the IS was identical for WASP-deficient cells and wt controls (Fig. 2,B). To rule out possible compensation during primary stimulation in culture, naive T cells were also tested. These, too, showed no actin defects at the IS (Fig. 2,C). Finally, labeling with anti-tubulin revealed that WASP-deficient T cells polarize their MTOC toward the bound APC as efficiently as wt T cells (Fig. 2 D). These data show that WASP is not required for either actin or MTOC polarization at the IS.

WASP-deficient T cells have no defect in actin or MTOC polarization at the IS. Previously activated (A, B, and D) or naive (C) T cells were conjugated for 10 min to CH12 B cells with or without 60 μg/ml DASP, fixed, and stained with rhodamine-phalloidin to visualize F-actin (A–C) or anti-tubulin to visualize the MTOC (D). A, Representative conjugates showing F-actin at the IS. B and C, quantitation of conjugates showing F-actin at the IS. D, Quantitation of conjugates showing the MTOC localized to the proximal third of the T cell. At least 50 conjugates were scored for each condition; results are from 1 representative experiment.
WASP-deficient T cells have no defect in actin or MTOC polarization at the IS. Previously activated (A, B, and D) or naive (C) T cells were conjugated for 10 min to CH12 B cells with or without 60 μg/ml DASP, fixed, and stained with rhodamine-phalloidin to visualize F-actin (A–C) or anti-tubulin to visualize the MTOC (D). A, Representative conjugates showing F-actin at the IS. B and C, quantitation of conjugates showing F-actin at the IS. D, Quantitation of conjugates showing the MTOC localized to the proximal third of the T cell. At least 50 conjugates were scored for each condition; results are from 1 representative experiment.
WASP is not required for protein organization within the IS
To test the role of WASP in SMAC formation, we localized talin and PKCθ in T-B conjugates. In AND Tg WASP-deficient T cells, as in wt T cells, PKCθ and talin localized to the IS, with PKCθ focused in the C-SMAC, and talin present in the surrounding P-SMAC (Fig. 3,A) Normal IS formation was also seen in WASP-deficient T cells on the DO11.10 Tg background (data not shown). Quantitative analysis revealed no defects in the overall efficiency with which these proteins accumulated at the IS (Fig. 3,B). Moreover, there were no defects in the frequency of PKCθ focusing within the C-SMAC; for both wt and WASP-deficient T cells, one-half of those conjugates showing PKCθ accumulation at the IS exhibited PKCθ focusing (data not shown). Because maintenance of an IS for at least 2 h is important to induce productive T cell activation (28), we asked whether WASP is required for IS maintenance. No differences in IS stability were observed for up to 180 min (Fig. 3 C). The ability to form a normal SMAC structure was not due to compensation in culture, because polarization of talin and PKCθ also proceeded normally in conjugates formed with naive WASP-deficient T cells (data not shown).

WASP-deficient T cells organize PKCθ and talin at the IS. Conjugates formed using previously activated T cells were double-labeled with anti-PKCθ and anti-talin Abs to analyze SMAC structure. A, Representative conjugates, with PKCθ in red and talin in green. Z-axis, Segregation of PKCθ and talin into C-SMAC and P-SMAC regions, respectively. B and C, Quantitation of conjugates showing PKCθ and talin polarization to the IS at 10 min (B) or up to 180 min (C). At least 50 conjugates were scored for each condition; results are from 1 representative experiment.
WASP-deficient T cells organize PKCθ and talin at the IS. Conjugates formed using previously activated T cells were double-labeled with anti-PKCθ and anti-talin Abs to analyze SMAC structure. A, Representative conjugates, with PKCθ in red and talin in green. Z-axis, Segregation of PKCθ and talin into C-SMAC and P-SMAC regions, respectively. B and C, Quantitation of conjugates showing PKCθ and talin polarization to the IS at 10 min (B) or up to 180 min (C). At least 50 conjugates were scored for each condition; results are from 1 representative experiment.
WASP facilitates talin and PKCθ recruitment at low peptide doses
To determine whether subtle defects in SMAC formation would become apparent at lower doses of peptide, dose-response analysis was performed. For these experiments, we used the MCC88–103 full agonist peptide, which stimulates T cell proliferation at ∼10-fold higher doses than the superagonist DASP peptide used above (26). At the highest doses of MCC88–103, WASP-deficient T cells polarized PKCθ and talin as efficiently as wt controls (Fig. 4,A). However, with decreasing peptide dose, the frequency of PKCθ and talin polarization fell off more rapidly for WASP-deficient T cells. These effects were most striking for talin, where the response fell to less than one-half that of wt cells by 1–0.1 μM peptide. As an alternate means of testing the role of WASP in setting the threshold for IS formation, we used the weak agonist peptide MCC99R, which stimulates T cell proliferation at ∼100-fold higher doses than MCC88–103 (29). Whereas the weak agonist did not reveal a latent defect in the WASP-deficient T cells when compared with the full agonist (Fig. 4, A and B), the WASP-deficient T cells again showed a significantly reduced frequency of PKCθ and talin polarization at low peptide doses. Thus, WASP facilitates the efficient recruitment of proteins to the IS under suboptimal stimulation conditions.
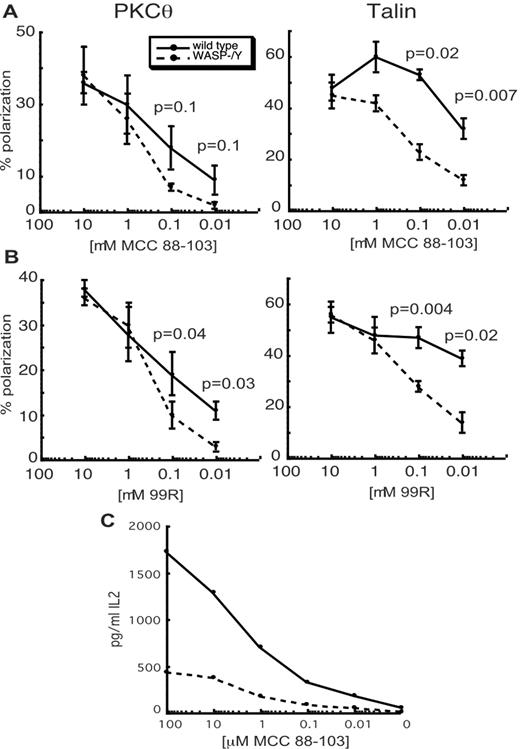
WASP-deficient T cells show defects in PKCθ and talin polarization at low doses of peptide but show IL-2 production defects at high doses. A and B, Experiments were performed as in Fig. 3 using the indicated doses of MCC88–103 peptide (A) or MCC99R peptide (B). Data are means ± SEM from 3 independent experiments with 50 conjugates each. p values were determined using the paired Student t test. C, Naive WASP+/Y or WASP−/Y lymph node T cells were incubated for 24 h with C3H splenocytes with the indicated concentrations of MCC88–103 peptide and analyzed by ELISA for IL-2 production. The result is from one representative experiment.
WASP-deficient T cells show defects in PKCθ and talin polarization at low doses of peptide but show IL-2 production defects at high doses. A and B, Experiments were performed as in Fig. 3 using the indicated doses of MCC88–103 peptide (A) or MCC99R peptide (B). Data are means ± SEM from 3 independent experiments with 50 conjugates each. p values were determined using the paired Student t test. C, Naive WASP+/Y or WASP−/Y lymph node T cells were incubated for 24 h with C3H splenocytes with the indicated concentrations of MCC88–103 peptide and analyzed by ELISA for IL-2 production. The result is from one representative experiment.
Comparison of the effects of WASP deficiency on IS formation and on IL-2 production shows a striking lack of correlation in these two phenotypes. Defects in IS formation were most apparent at the lowest doses of peptide tested, with no statistical difference from wt cells at 1 μM peptide or higher. In contrast, defects in IL-2 production were most pronounced at high doses of the DASP peptide (Fig. 1). To verify this distinction, we tested IL-2 production using the same peptides and doses used to measure IS formation. As shown in Fig. 4 C, WASP-deficient T cells showed pronounced IL-2 defects in response to APCs presenting the MCC peptide, with the largest defects observed at 1 μM and above. Similar results were obtained using MCC99R (data not shown). Thus, at the same peptide doses at which polarization of PKCθ and talin occurred normally, IL-2 production was severely impaired.
Discussion
It has been hypothesized that defects in IL-2 production in WASP-deficient T cells stem from defects in IS formation (1, 4, 5, 6). Surprisingly, however, we found that actin polymerization and MTOC polarization toward the IS occur normally in WASP-deficient T cells. Furthermore, segregation of PKCθ and talin into IS subdomains occurs appropriately in the absence of WASP. However, at low peptide doses, polarization of PKCθ and talin occurred less efficiently than in wt T cells. These results show that although WASP is not required for IS formation, WASP can lower the threshold for organizing these proteins at the IS. Importantly, IL-2 production was defective at all peptide doses, even those in which IS formation proceeded normally. These findings thus uncouple the effect of WASP on IS formation and IL-2 production and suggest that WASP function may be more complex than previously believed.
Although we cannot formally rule out that there are subtle defects in IS cytoskeletal architecture in the absence of WASP, we observed no difference in the shape of the cell-cell contact site or in the intensity of F-actin labeling. Thus, we conclude that WASP is not required for actin polymerization at the IS. In keeping with this finding, we previously reported that WASP-deficient T cells efficiently form conjugates with Ag-specific B cells (23). Moreover, although they did not directly examine actin responses in T-B conjugates, Krawczyk et al. (22) showed normal conjugation efficiency, normal actin capping, and normal LFA-1 responses in WASP−/− T cells responding to peptide Ag. The apparent discrepancy between these studies and previous work based on Ab stimulation may be explained by the additional complexities inherent in T-APC interactions, including the contribution of multiple costimulatory molecules. Indeed, there is evidence for complex feedback interactions in shaping normal cytoskeletal structure at the IS (25, 30). Additional study will be required to identify the proteins responsible for driving actin polymerization in the absence of WASP. Other WASP family members or proteins such as cortactin or formins could participate. Given the more severe actin phenotype of Vav−/− T cells, it seems likely that other Vav effectors such as Pak also affect actin dynamics.
Our findings differ from those of Badour et al. (15, 17), who found that OT-II TCR Tg WASP-deficient T cells were unable to polarize PKCθ to the IS in response to an Ag-pulsed B cell hybridoma. Unlike our results with AND TCR Tg and DO11.10 Tg T cells (23), or those of Krawczyk et al. using P14 TCR Tg T cells (22), the OT-II T cells failed to form conjugates efficiently, failed to polymerize actin normally, and therefore failed to organize a mature immune synapse architecture. The basis for the differences in conjugate formation and actin responses is unclear. One possibility is that there are weaker integrin or costimulatory interactions in the OT-II system. Another difference could stem from differences in T cell development and selection of transgene positive T cells in these different backgrounds. Regardless of the explanation, the AND TCR Tg system permits us to address the role of WASP in IS formation under conditions in which conjugate formation proceeds normally. On the basis of our findings in this system, we conclude that although WASP contributes to IS formation, WASP is not required for this process.
WASP-deficient T cells show defects in IL-2 production under conditions in which no defects in actin, PKCθ, or talin polarization are seen. Thus, the requirement for WASP in signaling IL-2 is not directly attributable to effects on these aspects of IS formation. These data fit well with recent evidence showing that IS formation most likely does not contribute to early TCR signaling but instead is necessary for dampening TCR activation through receptor down-regulation (31). It remains possible that WASP functions to regulate IL-2 production by controlling lipid raft-dependent signaling events (10). We were unable to analyze lipid raft dynamics in our system, due to high labeling of the B cells with FITC-cholera toxin. Alternatively, WASP may regulate IL-2 production via interactions with proteins such as WIP and Nck, which themselves interact with key signaling molecules, including PKCθ, ZAP-70, CrkL, SLP-76 and Vav (20, 32). In support of this idea, a WASP mutant that cannot bind WIP fails to induce NFAT activation (33), and overexpression of the WH1 domain of WASP affects proliferation without affecting cytoskeletal rearrangement (34). Finally, tyrosine phosphorylation of WASP could transduce signals downstream of WASP, possibly independently of actin regulation. Although additional study will be required to test the role of WASP as an adaptor protein, our findings clearly indicate that WASP does not function simply as a regulator of actin-dependent IS formation.
Acknowledgements
We thank Brian Evavold (Emory University, Atlanta, GA) for the MCC and 99R peptides, Shirley Bond in the University of Chicago Cancer Research Center Microscopy Facility (Chicago, IL), Renell Morgan for assistance with image analysis, and Anne Sperling and members of the Burkhardt laboratory for helpful discussions.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This work was supported by National Institutes of Health Grant R01-AI44835 (to J.K.B.).
Abbreviations used in this paper: IS, immune synapse; C-SMAC, central supramolecular activation cluster; P-SMAC, peripheral supramolecular activation cluster; MTOC, microtubule organizing center; WAS, Wiskott-Aldrich syndrome; WASP, WAS protein; WIP, WASP-interacting protein; Tg, transgenic; wt, wild type; MCC, moth cytochrome c; WH1, WASP homology 1.
